Publications
2025

F. Ye, S. Zhang, F. Lang, M. Raoufi, J. Liang, I. Levine, H. Hempel, D. Menzel, F. Zu, S. Albrecht, L. Korte, C. Messmer, J. Schön, S.W. Glunz, T. Unold, N. Koch, D. Neher, D. Ye, Y. Wu, M. Stolterfoht, "Minimizing Recombination at the Perovskite/C 60 Interface through a Volatile Highly Dense Molecular Interlayer", ACS Energy Letters 10, 2942 (2025), DOI: 10.1021/acsenergylett.5c00615
Advancing inverted perovskite solar cells requires effective strategies to mitigate nonradiative recombination at the perovskite/C60 interface. Here, we report a volatile material that forms a thin, dense interlayer that essentially eliminates the C60-induced nonradiative interfacial recombination loss despite not directly passivating the perovskite surface. Ultraviolet photoelectron spectroscopy highlights that the molecule forms a positive dipole layer on the surface that aligns the perovskite and C60 energy levels for electron conduction. Furthermore, the molecule’s volatile nature allows the use of a high-concentration solution that enables a high surface coverage (likely >99%) without increasing the thickness. The combination of these two effects yields an effective approach to suppressing interface recombination. The resulting triple cation perovskite solar cells achieved a power conversion efficiency of >25% and the devices maintain >90% of their initial efficiency after 1200 h of operation. Furthermore, the molecule is broadly applicable to various perovskite compositions and bandgaps.
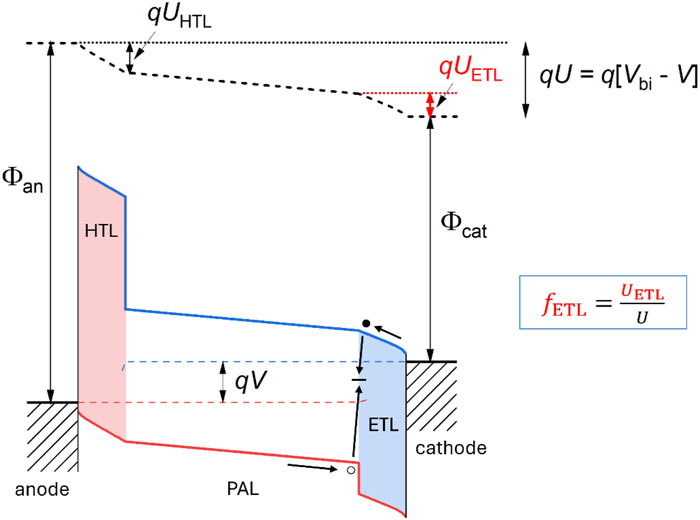
O.J. Sandberg, M. Kumar, M. Stolterfoht, D. Neher, A. Armin, "Analytical model for interface recombination limited ideality factors in p - i - n perovskite solar cells", APL Energy 3 (2025), DOI: 10.1063/5.0286898
Perovskite solar cells have emerged as a promising thin-film solar cell technology. However, compared to state-of-the-art photovoltaic technologies, perovskite solar cells still suffer from large nonradiative recombination losses. A dominant nonradiative recombination channel is recombination taking place at the interfaces between the perovskite active layer and the charge carrier transport layers. In this work, the influence of nonradiative interface recombination on the open-circuit voltage and the ideality factor in p-i-n type perovskite solar cells is clarified. We derive analytical expressions for the open-circuit voltage and the ideality factor. The analytical framework is substantiated by numerical drift-diffusion simulations. For devices dominated by interface recombination at the perovskite/transport layer interface, it is found that the open-circuit voltage is limited by the electrical potential loss across the charge transport layer. As a result, the corresponding ideality factor becomes dependent on the dielectric properties of the charge transport layers. These findings suggest that the voltage loss and ideality factor can be reduced by manipulating the thickness and dielectric properties of the charge transport layers.

M.S. Shadabroo, N. Tokmoldin, A. Shukla, A. Patterson, T.M. Melody, O. Alqahtani, B.A. Collins, D. Neher, S. Shoaee, "When the Triplet State Doesn't Matter: Insights into Its Impact on VOC", ACS Energy Letters 10, 2419 (2025), DOI: 10.1021/acsenergylett.5c00384
Organic solar cell efficiency, exceeding 20%, is limited by recombination losses from singlet and triplet charge-transfer (CT) states and local triplet excitons, impacting open-circuit voltage (VOC). Using PM6:o-IDTBR, a very low nonradiative voltage loss, ΔVnr = 160 mV, is achieved, despite the presence of triplet excitons (6 × 1015 cm–3). In employing the present system as a model exemplar, we elucidate the circumstance wherein, if the triplet lifetime surpasses the lifetime of the CT decay, the dissociation of triplet excitons to the CT state emerges as a feasible process. This, in turn, serves to reduce the manifestation of an additional loss channel from the T1 state and minimizes losses from T1. In PM6:o-IDTBR, the long triplet lifetime (10 μs) enables dissociation of the triplet state and limits the triplet-mediated recombination to ∼10%.

D. Neher, "Asymmetric side-chains work", Nature Materials 24, 338 (2025), DOI: 10.1038/s41563-024-02102-9
A new series of non-fullerene acceptors with asymmetric branched alkyl chains are developed to achieve more than 20% efficiency organic solar cells.

E. Gutierrez-Partida, M. Rusu, F. Zu, M. Raoufi, J. Diekmann, N. Tokmoldin, J. Warby, D. Menzel, F. Lang, S. Shah, S. Shoaee, L. Korte, T. Unold, N. Koch, T. Kirchartz, D. Neher, M. Stolterfoht, "Toward Understanding the Built-in Field in Perovskite Solar Cells through Layer-by-Layer Surface Photovoltage Measurements", ACS Applied Materials and Interfaces 17, 11176 (2025), DOI: 10.1021/acsami.4c14194
The built-in voltage (VBI) is a key parameter for solar cell operation, yet in perovskite solar cells the distribution, magnitude, and origin of the VBI remains poorly understood. In this work, we systematically studied the VBI in pin-type perovskite solar cells based on different hole transport layers (TLs). To this end, we determine the surface photovoltage (SPV) of partial and complete device stacks layer-by-layer by measuring the work function (WF) under dark and light (equivalent AM1.5G) conditions with Kelvin probe (KP) and photoemission spectroscopy (UPS) measurements in 3 different laboratories. We demonstrate that the SPV increases upon the addition of each additional layer until it equals the open-circuit voltage (VOC) of the full device. This suggests that both the electron and hole transport layer (HTL/ETL) enlarge the SPV, by improving the separation of photogenerated carriers. Yet, the contribution of both transport layers to the total SPV of the device is small (in the range of ≈100 to 200 meV) and the largest contribution to the SPV originates from the top metal electrode (≈500 meV). The results suggest that the VBI of pin-type perovskite solar cells is largely a result of the work-function difference of the electrodes. With regard to films (or incomplete cell stacks), our simulations can reproduce the measured SPV, and measured quasi-Fermi level splitting (>VOC) in partial cell stacks without a significant internal field consistent with the experimental data. This work establishes layer-by-layer SPV measurements, which are easily accessible, as a key tool for understanding device performance and internal energetics, similar to layer-by-layer QFLS measurements.

J.M. Cuervo-Ortiz, J.C.G. Palomares, S. Ozen, M. Härtel, S. Sarisozen, A. Dittwald, G. Kourkafas, A.-F. Castro-Méndez, F. Peña-Camargo, B.A. Seid, J. Bundesman, A. Denker, H.-C. Neitzert, D. Neher, E. Stoll, S. Linke, F. Lang, "Moon photovoltaics utilizing lunar regolith and halide perovskites", Device, 100747 (2025), DOI: 10.1016/j.device.2025.100747
Powering future Moon settlements requires reliable and cost-effective energy generation with high specific power. Here, we propose halide perovskite photovoltaics (PV) fabricated on regolith-based moonglass that could be produced on the Moon, thereby saving 99% of material transport weight. This enables effective specific power ratios, over 22–50 W/g, a factor of 20–100 higher compared to traditional space PV solutions, while not compromising radiation shielding, reliability, and mechanical stability as done until now. Using anorthosite high-glass-forming regolith simulant, we achieve transparent moonglasses that allow depositing high-quality perovskites. We achieve performances on par with references, revealing the potential of perovskite-based Moon photovoltaics, and propose routes to achieve power conversion efficiencies of 23%. The moonglass exhibits high tolerance to high-energetic proton irradiation, which, when combined with the radiation tolerance of perovskites, allows highly radiation-tolerant, reliable devices paving the way to future sustainable lunar-energy solutions.

B.A. Seid, S. Ozen, A.-F. Castro-Méndez, D. Neher, M. Stolterfoht, F. Lang, "Mitigating Mobile-Ion-Induced Instabilities and Performance Losses in 2D Passivated Perovskite Solar Cells", Advanced Materials, 2501588 (2025), DOI: 10.1002/adma.202501588
Bulky ammonium salt-based passivation is an effective strategy for enhancing the performance and stability of perovskite solar cells (PSCs). Especially, phenethylammonium iodide (PEAI) is known to greatly improve open-circuit voltage (VOC) and fill factor (FF). Despite these benefits, PEAI passivation leads to substantial short-circuit current density (JSC) losses and rapid degradation under operational conditions. In this work, it is revealed that the JSC loss as well as the accelerated degradation in PEAI-passivated devices is caused by an increased mobile ion density. To mitigate this performance and stability-limiting mechanism, ultrathin layers of ammonium benzenesulfonate (ABS) and/or ethylenediammonium diiodide (EDAI2) salts are then introduced between the PEAI and the perovskite, which stabilize the 2D perovskite layer and impede diffusion even under upon prolonged illumination. This leads to a reduced mobile ion density both in fresh devices and in the long term, lowering losses JSC, and thus enables power conversion efficiencies of ≈25% with enhanced stability. Overall, this study not only addresses the limitations of PEAI-based 2D passivation but also paves the way for understanding 2D-induced ionic JSC losses.

C. Wang, R.C.I. MacKenzie, U. Würfel, D. Neher, T. Kirchartz, C. Deibel, M. Saladina, "Transport Resistance Dominates the Fill Factor Losses in Record Organic Solar Cells", Advanced Energy Materials (2025), DOI: 10.1002/aenm.202405889
Organic photovoltaics (OPV) are a promising solar cell technology well-suited to mass production using roll-to-roll processes. The efficiency of lab-scale solar cells has exceeded 20% and considerable attention is currently being given to understanding and minimizing the remaining loss mechanisms preventing higher efficiencies. While recent efficiency improvements are partly owed to reducing non-radiative recombination losses at open circuit, the low fill factor (FF) due to a significant transport resistance is becoming the Achilles heel of OPV. The term transport resistance refers to a voltage and light intensity-dependent charge collection loss in low-mobility materials. In this perspective, it is demonstrated that even the highest efficiency organic solar cells (OSCs) reported to-date have significant performance losses that can be attributed to transport resistance and that lead to high FF losses. A closer look at the transport resistance and the material properties influencing it is provided. How to experimentally characterize and quantify the transport resistance is described by providing easy to follow instructions. Furthermore, the causes and theory behind transport resistance are detailed. In particular, the relevant figures of merit (FoMs) and different viewpoints on the transport resistance are integrated. Finally, we outline strategies that can be followed to minimize these charge collection losses in future solar cells.

A. Shukla, M. Pranav, G. He, J.T. Blaskovits, D. Mascione, Y. Cao, Y. Gong, D.B. Riley, J.A. Steele, E. Solano, A. Ehm, M.S. Shadabroo, A. Armin, S. Shoaee, D.R.T. Zahn, Y. Li, L. Meng, F. Lang, D. Andrienko, D. Neher, "Discerning Performance Bottlenecks of State‐of‐the‐Art Narrow Bandgap Organic Solar Cells", Advanced Energy Materials (2025), DOI: 10.1002/aenm.202502398
Discerning loss mechanisms in organic solar cells with narrow optical bandgap is critical for the development of conventional and next-generation photovoltaic technologies, especially for tandem and semi-transparent solar cells. Here, all photocurrent losses are quantitatively deconvoluted in two low-bandgap (Eg≈1.23 eV) binary systems using structurally analogous non-fullerene acceptors (NFAs), namely BTPV-4F-eC9 and BTPV-4Cl-eC9. Bias-dependent free charge generation and photoluminescence studies pinpoint geminate charge transfer (CT) state recombination as the predominant photocurrent limitation in both systems, compared to parent Y6-blends. Transient absorption spectroscopy too reveals a critical competition between CT decay and separation dynamics. Theoretical calculations uncover multiple stable molecular conformers that restrict NFA aggregation, aligning with morphological studies, resulting in poor CT separation in photoactive blends. Owing to CT loss pathways, free charge recombination in both low-bandgap systems is closer to the Langevin limit than in PM6:Y6. Nonetheless, they exhibit overall voltage losses of ≈0.56 V comparable to PM6:Y6, and efficient exciton dissociation despite a lower driving force. Current–voltage simulations show that suppressing geminate losses can vitally balance recombination pathways to unlock photocurrent potential of low-bandgap blends. Further optimization of the charge carrier mobility would push the PCE >16%, moving the internal quantum efficiency toward the detailed balance limit.

S. Chandrabose, A.M. Valencia, M. Raoufi, N. Alshehri, T.M. Clarke, F. Laquai, C. Cocchi, D. Neher, "Mitigating triplet loss in 2D WSe2/non-fullerene heterostructures using halogenated acceptors", Materials Horizons (2025), DOI: 10.1039/d4mh00894d
Two-dimensional transition metal dichalcogenides (2D TMDCs) can be combined with organic semiconductors to form hybrid van der Waals heterostructures. Specially, non-fullerene acceptors (NFAs) stand out due to their excellent absorption and exciton diffusion properties. Here, we couple monolayer tungsten diselenide (ML-WSe2) with two well performing NFAs, ITIC, and IT-4F (fluorinated ITIC) to achieve hybrid architectures. Using steady state and time resolved spectroscopic techniques, we reveal sub-picosecond free charge generation in the heterostructure of ML-WSe2 with ITIC, where however, bimolecular recombination of spin uncorrelated charge carriers with possible contributions from geminate charge recombination cause rapid formation of low-lying triplet (T1) states in ITIC. Importantly, this unwanted process is effectively suppressed when the fluorinated derivative of ITIC, IT-4F, is deposited on ML-WSe2. We observe a similar scenario when replacing the ML-TMDC with copper thiocyanate (CuSCN) as the hole acceptor meaning that triplet state formation is not driven by the spin–orbit coupling of ML-WSe2. From ab initio calculations based on density functional theory, we interpret the high triplet formation in the ML-WSe2/ITIC hybrid bilayer due to changes in the nature and energies of the interfacial charge transfer (CT) levels. Our results highlight the delicate balance between excitons and charges in such inorganic/NFA heterostructures.