Articles
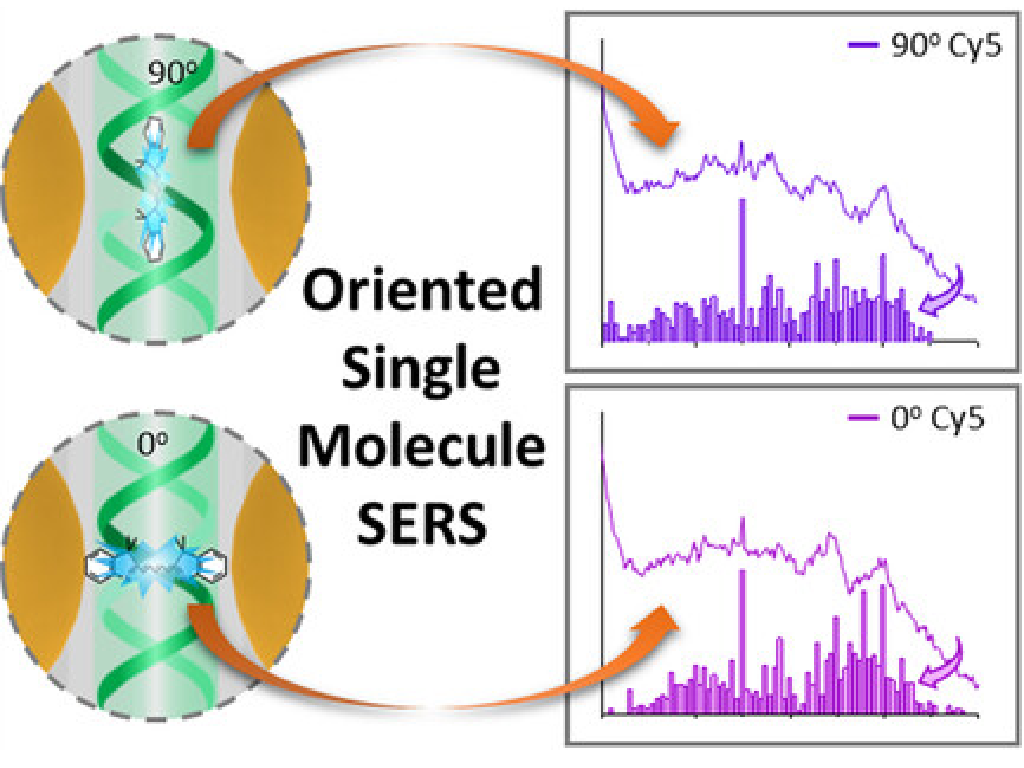
Y. Kanehira, A. K. Adamczyk, E. Titov, P. Saalfrank, K. Kołątaj, G. P. Acuna, S. Kogikoski, and I. Bald
Molecular Orientation Effects on Surface-Enhanced Raman Scattering Spectra at the Single-Molecule Level Revealed by DNA Origami Plasmonic Antennas
Small Structures, 2025, 0, e202500692
Characterizing single-molecule signals in surface-enhanced Raman scattering (SERS) remains challenging, particularly in identifying the sources of spectral fluctuations. While prior studies focused on nanoparticle composition and surface restructuring, they often overlook the molecule's behavior, partly due to limitations in controlling molecular positioning or the complexity introduced by bianalyte methods. Here, we employ theoretical methods to model the Raman spectra of a Cy5 dye, demonstrating that spectral variations occur when the molecule aligns parallel to the external electric field. By leveraging DNA's capability to immobilize and orient molecules, we experimentally acquired single-molecule SERS spectra for dye molecules in parallel and perpendicular orientations to the DNA double helix. Notably, distinct spectral differences emerged between these orientations, suggesting that factors like functional group proximity to nanoparticle surfaces and the electric-field intensity within nanocavities critically impact SERS outcomes. Density functional theory calculations further elucidate these effects, accounting for finite-field effects, molecular orientation, and surface interactions. These findings enhance our understanding of single-molecule behavior within plasmonic hot spots, offering new insights into the role of molecular orientation in SERS and informing future nanotechnology applications.
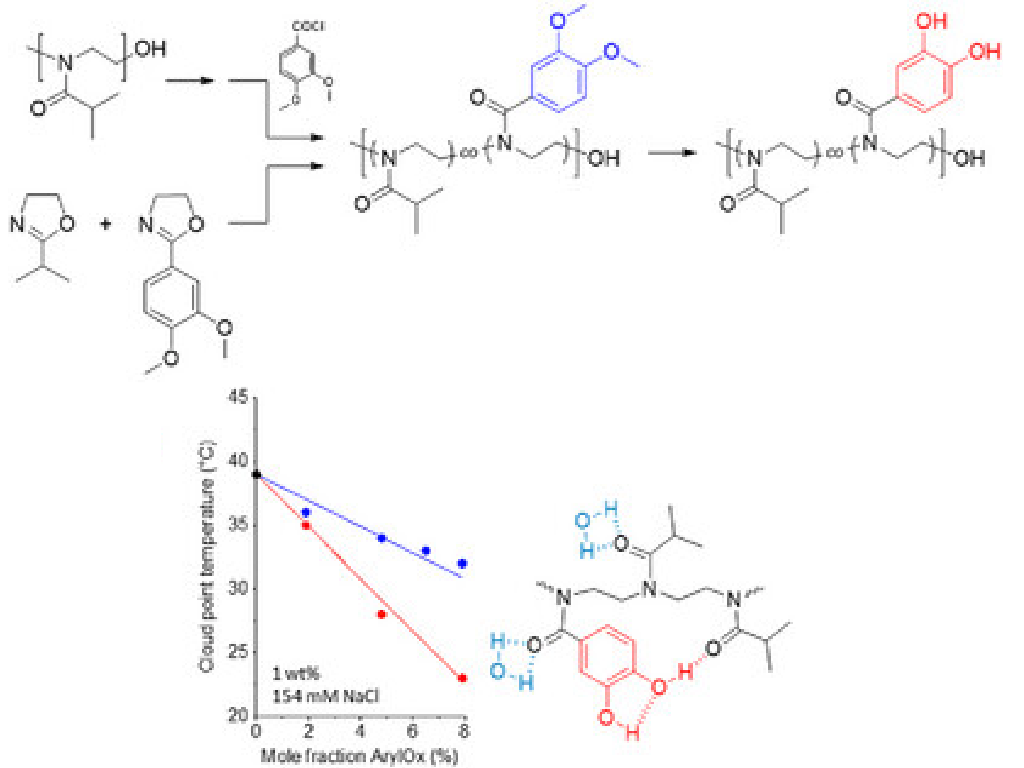
N. Madaj, N. Lüdecke, S. Kogikoski Jr., I. Bald, and H. Schlaad
Catechol-Containing Poly(2-isopropyl-2-oxazoline): Synthesis and Thermoresponsive Behavior in Aqueous Salt Solutions
Macromolecular Rapid Communications (2025): e00719
Catechol-containing poly(2-isopropyl-2-oxazoline)s (PiPOx) were obtained either by functionalization of partially hydrolyzed PiPOx with 3,4-dimethoxybenzoyl chloride or by microwave-assisted cationic ring-opening copolymerization of 2-isopropyl-2-oxazoline with 2-(3,4-dimethoxyphenyl)-2-oxazoline, followed by quantitative deprotection of methylated catechols. All synthesized polymers were soluble in aqueous salt solutions at room temperature and showed thermoresponsive LCST behavior. The cloud point temperature (TCP ) remained virtually unchanged in the presence of different cations, while it was strongly affected by chaotropic and kosmotropic anions, which is in line with the Hofmeister series. Intriguingly, TCP was lower for the catechol-containing PiPOx as compared to the protected precursor copolymer and PiPOx, which is rationalized by hydrogen bonding interactions between catechol and tertiary amide units of the polymer backbone, as indicated by Raman spectroscopy, decreasing the hydration level of the polymer.

N. Jahn, E. Titov, and P. Saalfrank
Ground state quantum chemistry, excited state dynamics, and time-resolved x-ray absorption spectroscopy of substituted thiophenols
The Journal of Chemical Physics 163, 174304 (2025)
Time-resolved x-ray absorption spectroscopy is at present rapidly gaining popularity as a powerful analytic technique for studying electronically excited molecules. The method effectively combines the site- and element-specific resolution of x rays with the capability of probing both optically bright and dark states on the femtosecond time scale. However, the high information content in corresponding experimental data usually requires support from theoretical simulations for interpretation. In the present study, we combine trajectory surface hopping (TSH) dynamics, including spin–orbit coupling, with transition potential/Δ-Kohn–Sham x-ray absorption simulations based on density functional theory to simulate time-resolved x-ray absorption spectra of 4-nitrothiophenol (4-NTP), a system well known in the field of plasmonic chemistry and catalysis. We find that 4-NTP in the gas phase undergoes rapid internal conversion via low-lying, dark nπ* states of the NO2-group after excitation to the first bright ππ* state, which can be directly tracked in the time-resolved C1s and O1s x-ray absorption spectra. Furthermore, picosecond intersystem crossing in photoexcited 4-NTP is identified, which agrees with supporting TSH simulations based on second-order algebraic-diagrammatic construction [ADC(2)] theory. We also discuss a variety of ground state quantum chemical and static UV/Vis spectroscopic properties of 4-NTP in comparison to the structurally similar 4-halogenated thiophenols (4-HAT or 4-XTP), which we aim to investigate in greater detail in a future study.
P. A. Albrecht, E. W. Fischer, T. Klamroth, P. Saalfrank
High-harmonic generation under electronic strong coupling: A time-dependent combined quantum electrodynamics/quantum chemistry study
The Journal of Chemical Physics 163, 164302 (2025)
The creation of light–matter hybrid states, polaritons, in a cavity offers new intriguing opportunities to manipulate the electronic structure and electron dynamics of atoms and molecules. Here, we investigate the effect of strong electronic coupling between atoms or molecules and field modes of a Fabry–Pérot cavity on High-Harmonic Generation (HHG) spectra within a theoretical model study. We assume that the atom or molecule is driven by an intense classical laser field, giving rise to HHG, while being strongly coupled to quantized cavity modes as described by the Pauli–Fierz Hamiltonian in the framework of molecular quantum electrodynamics. Specifically, as a test case, we first consider a model Hamiltonian of a one-dimensional hydrogen atom coupled to a cavity mode, which can be treated “numerically exact” using grid methods. Furthermore, a hydrogen molecule coupled to a cavity mode is considered and treated within a recently suggested QED-TD-CI (Quantum Electrodynamics Time-Dependent Configuration Interaction) method [Weidman et al., J. Chem. Phys. 160, 094111 (2024)]. The resulting HHG spectra show (i) a suppression of the harmonic cutoff in line with the excitation of quantum light in the cavity and, in some cases, (ii) enhancement of some harmonics of the coupled light–matter system.

R. Jutas, J. Grumm, Y. U. Staechelin, A. Pugžlys, A. Baltuška, A. Knorr, H. Lange
Nonlinear Transmission of Strong THz-Electric Fields Through Thin Gold Films
Advanced Optical Materials, 2025, e01831
This study reports on a nonlinear response in thin gold films in the terahertz (THz) frequency region. In an experimental study, the THz-electric peak field strength is increased and the transmission through the gold film is found to increase nonlinearly with the field strength when it exceeds a few hundred kV cm−1. Numerical calculations of the electron dynamics based on the momentum-resolved Boltzmann equation show that the fast ((sub-) picosecond) electron depolarization activates a THz field-induced Pauli blocking nonlinearity of the electron–phonon interaction, resulting in an increased transmission.
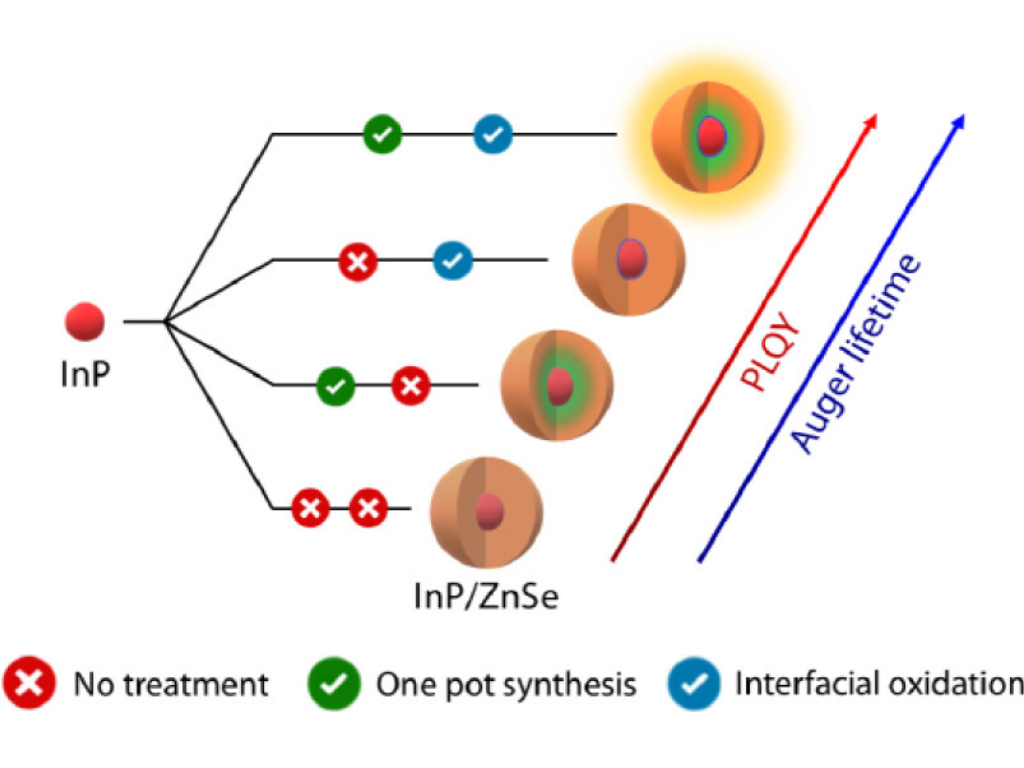
L. Giordano, P. Schiettecatte, Y. Coppel, Q. Zhao, Y. U. Staechelin, G. Bonifas, H. Van Avermaet, C. Nayral, H. Lange, A. Vantomme, F. Delpec, and Z. Hens
The Core/Shell Interface in InP/ZnSe Colloidal Quantum Dots
Chemistry of Materials 2025, 37, 21, 8724–8732
InP/ZnSe core–shell quantum dots (QDs) can emit spectrally narrow light with high efficiency, but the relation between the QD properties and the composition of the core–shell interface remains unclear. Here, we compare 4 different batches of InP/ZnSe QDs, formed with or without intermediate purification and InP surface oxidation before shell growth. Elemental analysis and solid-state NMR show that the presence of InCl3 during ZnSe shell growth leads to indium incorporation into the ZnSe shell, while interfacial oxidation creates a polyphosphate at the core/shell interface. The sample in which both interfacial engineering approaches were applied features a higher photoluminescence quantum yield and the slowest biexciton Auger recombination rate. These findings support emerging insights on the InP/ZnSe core/shell interface in the literature and pave the way for further improving the optoelectronic properties of these materials by adjusting the interfacial composition.
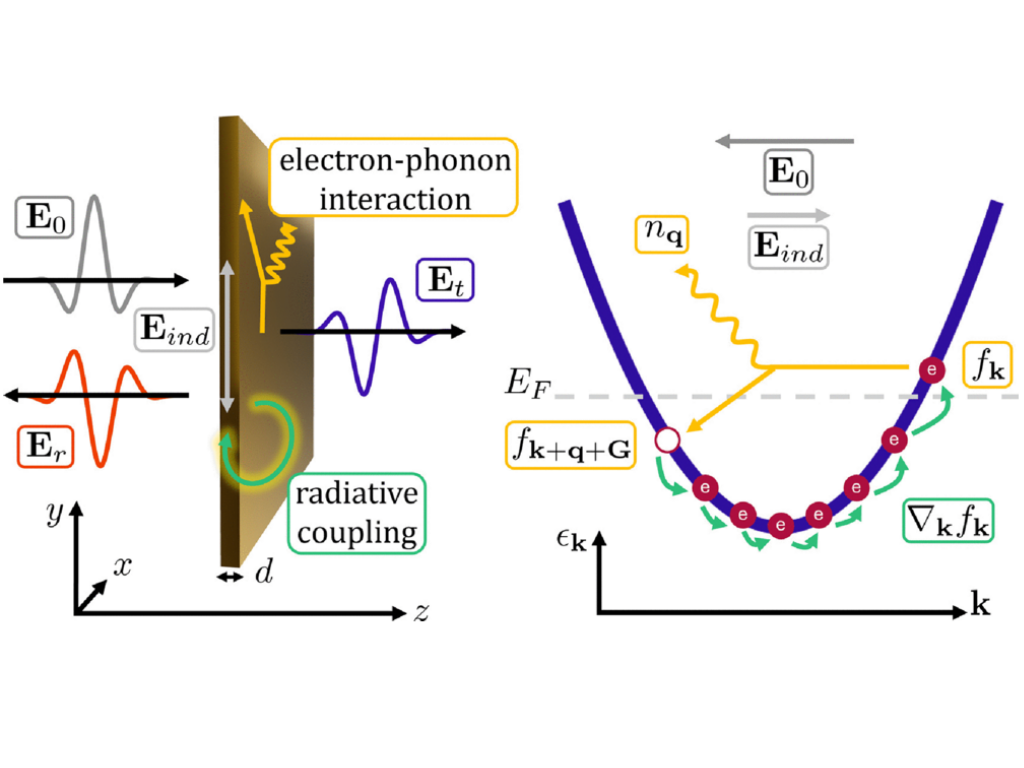
J. Grumm, M. Selig, H. Lange, and A. Knorr
Theory of nonlinear electron relaxation in thin gold films and their signatures in optical observables
Physical Review B 112, 245402 (2025)
Based on the momentum-resolved Boltzmann equation, we provide self-consistent numerical calculations of the dynamics of conduction electrons in thin noble metal films after linear and nonlinear optical excitations with infrared and terahertz frequencies. Focusing exclusively on electron-phonon interaction, orientational relaxation is introduced and acts as dephasing of the optical excitation on a scale of tens of fs. In the linear regime, our numerical results agree with the field-strength-independent orientational relaxation rate and correspondingly fits of experimental data to a Drude model and predict for nonlinear excitations a field-strength-dependent increase of the orientational relaxation rate. In the THz regime, where the orientational relaxation proceeds faster than the oscillation cycle of the excitation THz field, a new high-order dissipative Kerr-type nonlinearity is predicted. This nonlinearity originates from the Pauli blocking included in the electron-phonon scattering and results in a nonlinearly increasing transmission of the film, detectable in experiments.
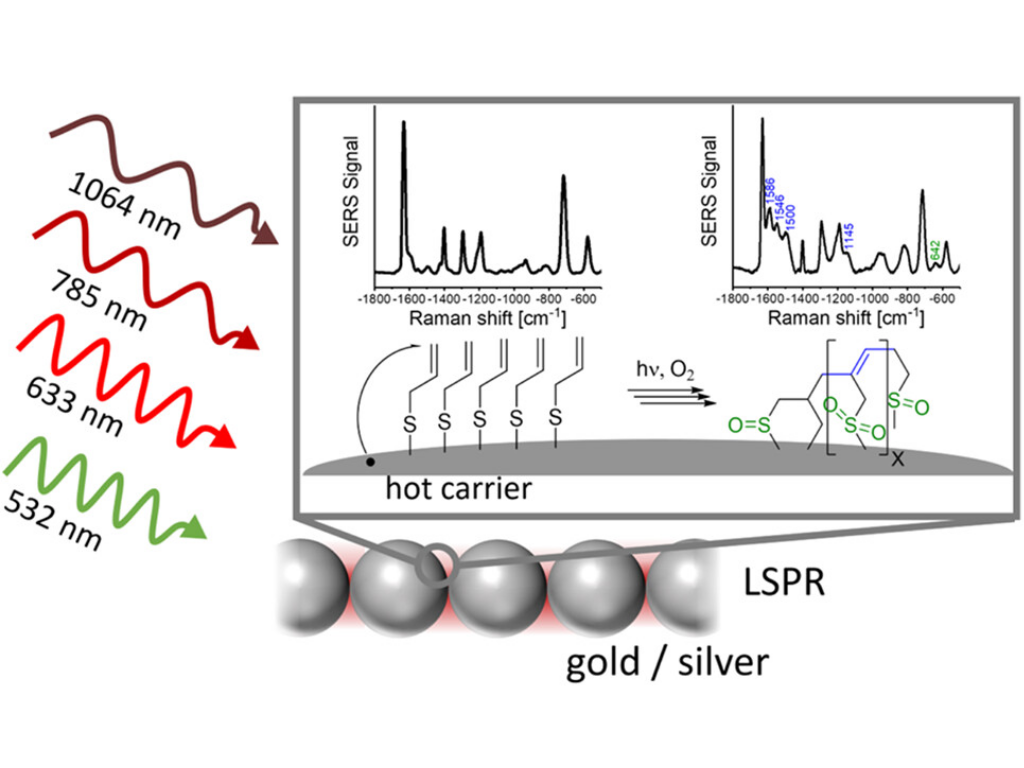
L. Dannenberg, H. Schlaad, and J. Kneipp
Hot-Carrier-Induced Transformation of Allyl Mercaptan on Silver and Gold Nanostructures
ACS Catalysis 2025, 15, 22, 19328–19342
The generation of hot charge carriers from plasmonic metals has been harnessed in a range of plasmon-catalyzed reactions. Here, we report the chemical reaction of allyl mercaptan (2-propene-1-thiol) on different plasmonic silver and gold substrates and elucidate the role of localized surface plasmon resonances in the reaction. The decay of localized plasmons can create hot carriers. Additionally, high local fields of the plasmonic nanoparticles at a matching energy range enhance directly optically excited interband transitions in metal nanoparticles. Moreover, the high local fields enable observation of surface-enhanced Raman scattering (SERS) and also nonlinear surface-enhanced hyper-Raman scattering (SEHRS) from the reaction product. We introduce monitoring of the reaction-related change based on the full spectral profile represented by the principal component scores rather than by individual spectral signals, and thereby can base the kinetic description on different reactant and product functional groups at the same time. As evidenced by the plasmon-enhanced vibrational spectra collected in the course of the reaction, allyl mercaptan forms alkyl oligomers with different chain lengths with oxidized sulfur groups. The direct observation of carbon–metal bond formation on silver and gold nanostructures and the dependence of the reaction rates on the excitation wavelength and intensity indicate an activation of the reaction by hot electrons. We propose that their interaction with the double bond initiates a radical oligomerization that is accompanied by a plasmon-supported elimination of hydrogen to enable the formation of unsaturated chains. A high reaction efficiency is observed when plasmon resonances fall in the energy range of interband transitions, as it occurs in some experiments conducted with gold nanoparticles, and when high local fields can enhance the production of hot carriers. The activation of double bonds on plasmonic surfaces can be used to produce functionalized nanostructures and materials.
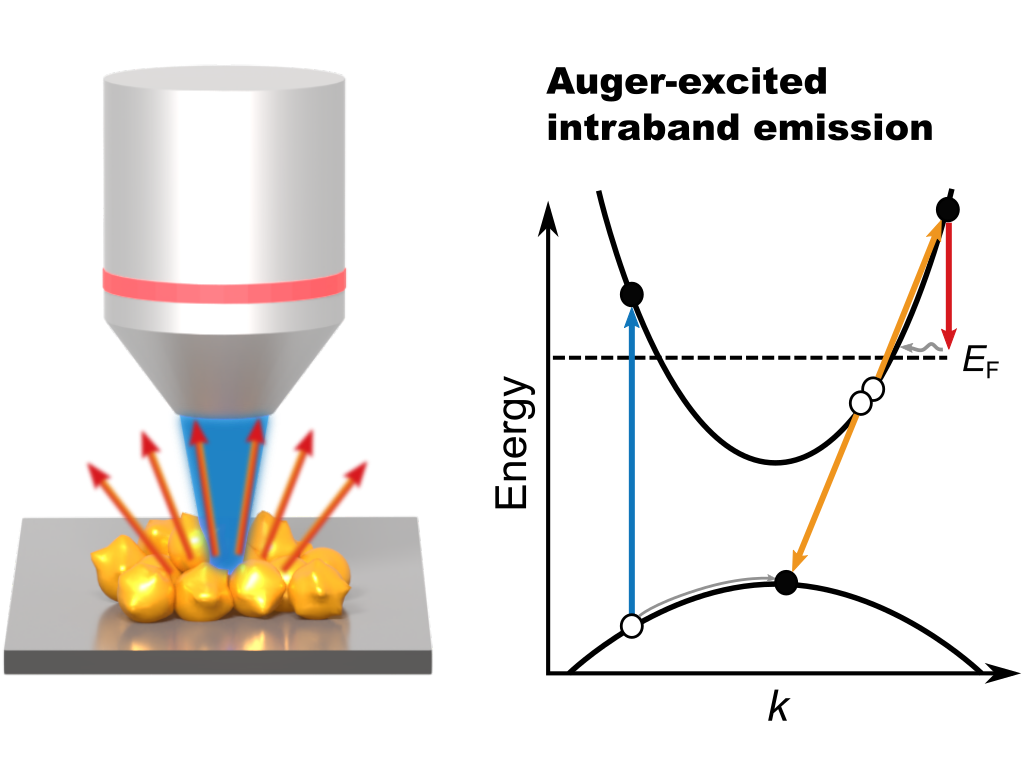
W. Koopman, J. Kutschera, F. Stete, and M. Bargheer
Auger-Excited Photoluminescence from Gold Nanoflowers
ACS Nano, 2025, 19, 39, 34517–34526
Photoluminescence from metal nanostructures offers a promising means of studying excited charge processes in metal nanostructures. Moreover, they have many potential applications in sensing, imaging, and nanothermometry. However, a general understanding of the emission from metal nanoparticles has not yet been achieved. In particular, the possible presence of sequential emission mechanisms involving the excitation of conduction band electrons via interband Auger scattering remains unclear. In this article, we provide spectroscopic evidence of Auger-excited intraband emission from gold nanoflowers. We employ a combination of photoluminescence and photoluminescence excitation spectroscopy to investigate the excitation pathways in films of gold nanoflowers. While, on the one hand, the excitation spectrum clearly demonstrates absorption by interband transitions, the emission spectra can be unequivocally assigned to intraband recombination. The combination of these two observations can be conclusively explained only by Auger-excited intraband emission. These results suggest Auger excitation to be a promising route to generate energetic nonthermal electrons with energies substantially above the Fermi level. Exploiting this effect could strongly benefit applications for nanoluminescent probes and the progress of plasmon catalysis.
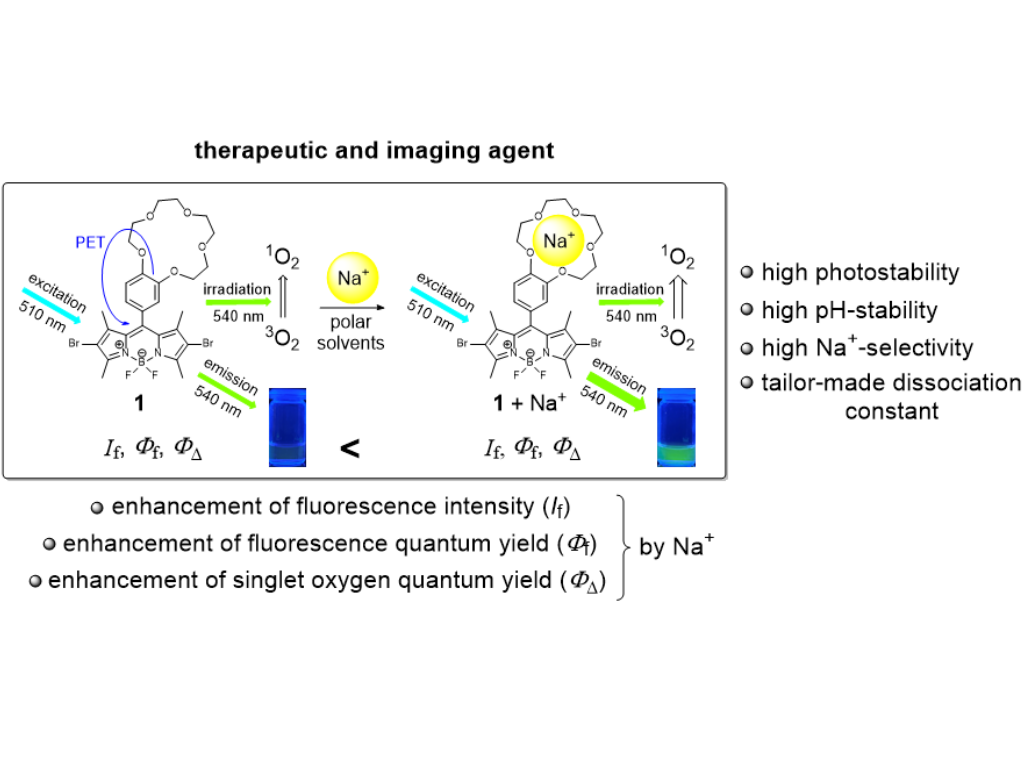
T. Schwarze, M. Al Akrami, J. Heinrich, V. Hergl, E. Sperlich, A. Kelling, T. Sprenger, N. Jahn, T. Klamroth, and N. Kulak
A sodium ion-selective photosensitizer: Dibrominated F-BODIPY as a fluorescence imaging and therapeutic agent
Physical Chemistry Chemical Physics, 2025, 27, 24178-24183
Herein, we report that the production of singlet oxygen (1O2) is exclusively regulated by sodium ions in aqueous solution by the use of a Na+-selective photosensitizer (PS), a 2,6-dibrominated F-BODIPY dye equipped with benzo-15-crown-5. The PS showed an enhanced fluorescence quantum yield (Φf) and an enhanced singlet oxygen quantum yield (ΦΔ) in the presence of Na+ caused by suppressing photoinduced electron transfer (PET). A detailed theoretical study uncovered the underlying photophysical pathways which are responsible for both functional characteristics of the PS, therapeutic and Na+ imaging properties.

F. Stete, S. Kesarwani, C. Ruhmlieb, S. H. C. Askes, F. Schulz, M. Bargheer, and H. Lange
Inverted temperature gradients in gold–palladium antenna-reactor nanoparticles
Nature Communications, 16, 8168 (2025)
In addition to enhanced fields and possible charge transfer, the concentration of photothermal energy at the nanoscale is a central feature of plasmon-driven photochemistry. It is well known that light energy can be efficiently concentrated in metal nanoparticles to length scales far below the wavelength of light. Here we demonstrate that the energy absorbed by a gold nanoparticle can be further localized within a bimetallic gold-paladium nanoparticle system by the dissipation of energy into the attached palladium satellite nanoparticles. After pulsed excitation of the gold core, the satellites collect nearly all photothermal energy and heat up by 180 K while the light-absorbing gold core remains much colder. By comparing transient absorption dynamics of a series of bimetallic nanoparticles with a three-temperature model, we can precisely assess the temperatures of the electronic and vibrational subsystems. We find a strong inverted temperature gradient that opposes the direction of energy input and concentrates the light energy at the active catalytic nanosite.
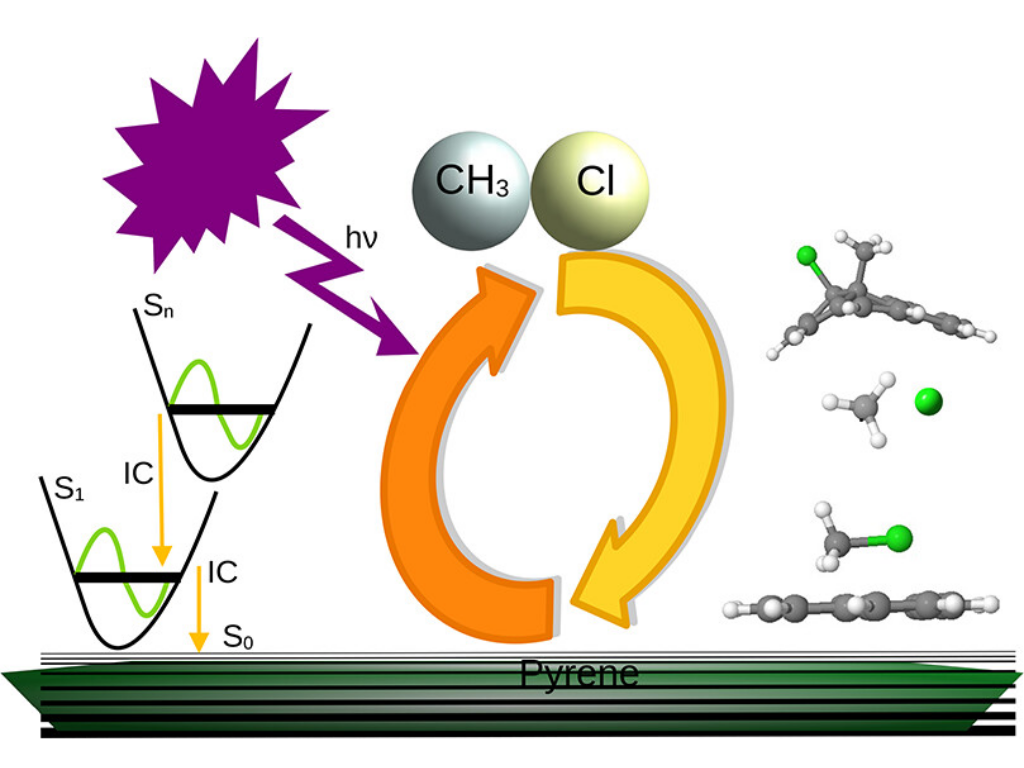
E. Mazarei, E. Titov, and P. Saalfrank
Surface Hopping Molecular Dynamics Simulations for Photochemistry Involving Pyrene and CH3Cl
Journal of Physical Chemistry A , 129, 31, 7102–7114, (2025)
Pure or halogenated polycyclic aromatic hydrocarbons (PAHs) and saturated halogenated hydrocarbons are both classes of harmful chemicals found in Earth’s atmosphere, often involved in photochemical reactions. On a positive side, the photoreaction of PAHs with halogenated hydrocarbons serves as a model and offers routes to functionalized nanostructured carbon-based materials with tailored optoelectronic properties. Mechanistic studies on the photoreactions of these chemicals, possibly with each other, are therefore clearly of interest but still comparatively rare. In the present work, as a representative case study, the photophysics (spectra, excited-state lifetimes) and photoreaction dynamics of van der Waals or chemically bound complexes of pyrene (C16H10) and methyl chloride (CH3Cl) were investigated using a combination of computational techniques, thereby delivering time- and atom-resolved information on postexcitation processes. Structural optimizations of possible reactants and products, as well as excited states and absorption spectra, were obtained by semiempirical (AM1) configuration interaction singles (CIS) and (time-dependent) density functional theory (DFT), respectively. Nonadiabatic surface hopping dynamics (NASH) based on AM1-CIS provided excited-state lifetimes and were used to explore various photochemical channels of CH3Cl physisorbed or covalently bound to pyrene.
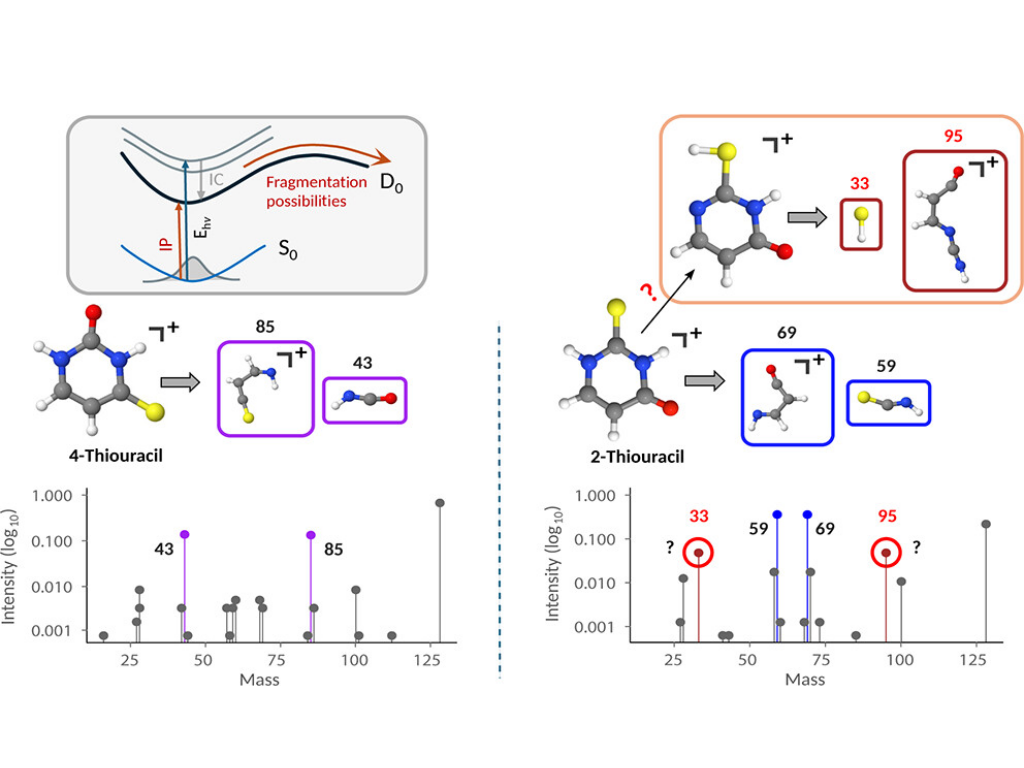
B. Roy, E. Titov, M. S. Robinson, M. Gühr, and P. Saalfrank
Dissociative Photoionization of 2-Thiouracil and 4-Thiouracil: A Molecular Dynamics Study
Journal of Physical Chemistry A, 129, 32, 7352–7364 (2025)
Thionated nucleobases have drawn considerable attention due to their role in medical treatment and biology, which triggered studies of their photophysics and photochemistry using various experimental and theoretical techniques. In particular, vacuum ultraviolet (VUV)-induced dissociative photoionization of 2-thiouracil (2-TU) has been recently studied using synchrotron radiation [Robinson et al., Molecules 2023, 28, 2354]. Here, using molecular dynamics simulations at the semiempirical OM2 level, we study the fragmentation dynamics of the cations of 2-TU and its isomer, 4-thiouracil (4-TU). Considering various VUV photon energies, we calculate energy-resolved mass spectra and breakdown diagrams for 2-TU and 4-TU. Remarkably, we find that the major fragments are different between the two compounds: 69 amu (C3NH3O+) for 2-TU and 85 amu (C3NH3S+) for 4-TU. Our simulations provide direct mechanistic insight into this observation and the fragmentation processes of thionated uracils in general.
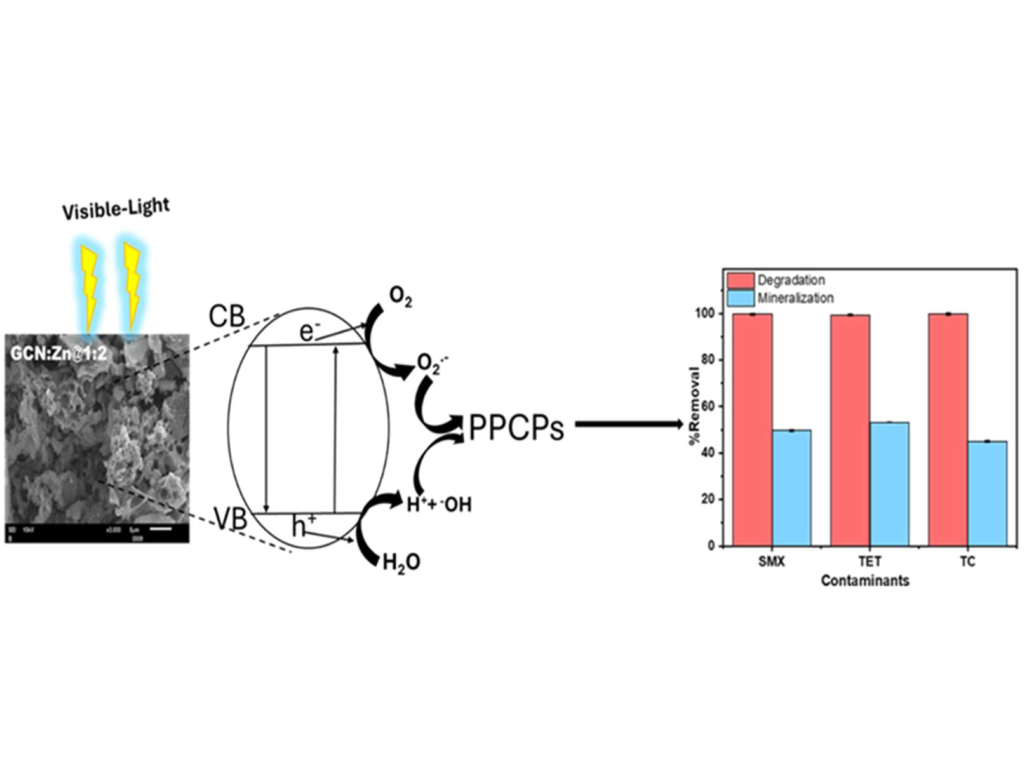
C. G. Olorunnisola, D. Olorunnisola, C. Neumann, W. Koopman, C. Günter, H. Seitz, H. M. Rawel, E. I. Unuabonah, and A. Taubert
Zn-Doped Porous Graphitic Carbon Nitride: A High-Performance Catalyst for the Photodegradation of Pharmaceuticals and Personal Care Products
ACS Omega 2025, 10, 36, 41395–41412
In this study, photocatalytically active graphitic carbon nitride (GCN) photocatalysts with varying amounts of zinc salts were synthesized using a solvent-free, scalable, and eco-friendly method. The photocatalysts were characterized using X-ray powder diffraction, scanning electron microscopy, nitrogen sorption, thermogravimetric analysis, UV–vis diffuse reflectance, and time-resolved photoluminescence (TRPL) spectroscopy. The addition of zinc to the GCN yields materials (GCN:Zn) with an enhanced surface area up to ca. 150 m2/g. Moreover, Zn incorporation affects the electronic structure of GCN and improves the electron transfer rate in the materials. The GCN:Zn materials show significantly improved photocatalytic activity for the degradation of pharmaceutical and personal care products (PPCPs) such as sulfamethoxazole (SMX), tetracycline (TET), and triclosan (TC) compared to zinc-free GCN. Although the degradation efficiency exceeds 98%, mineralization of the PPCPs is moderate (45–53%) due to the formation of stable intermediates. The treated effluents are nontoxic to Escherichia coli and Staphylococcus xylosus, indicating that no harmful intermediates form during the photocatalytic degradation of the PPCPs. The GCN:Zn photocatalysts demonstrate excellent reusability and stability for the degradation of PPCP over four cycles with minimal loss in activity, showcasing their potential for sustainable water treatment applications.
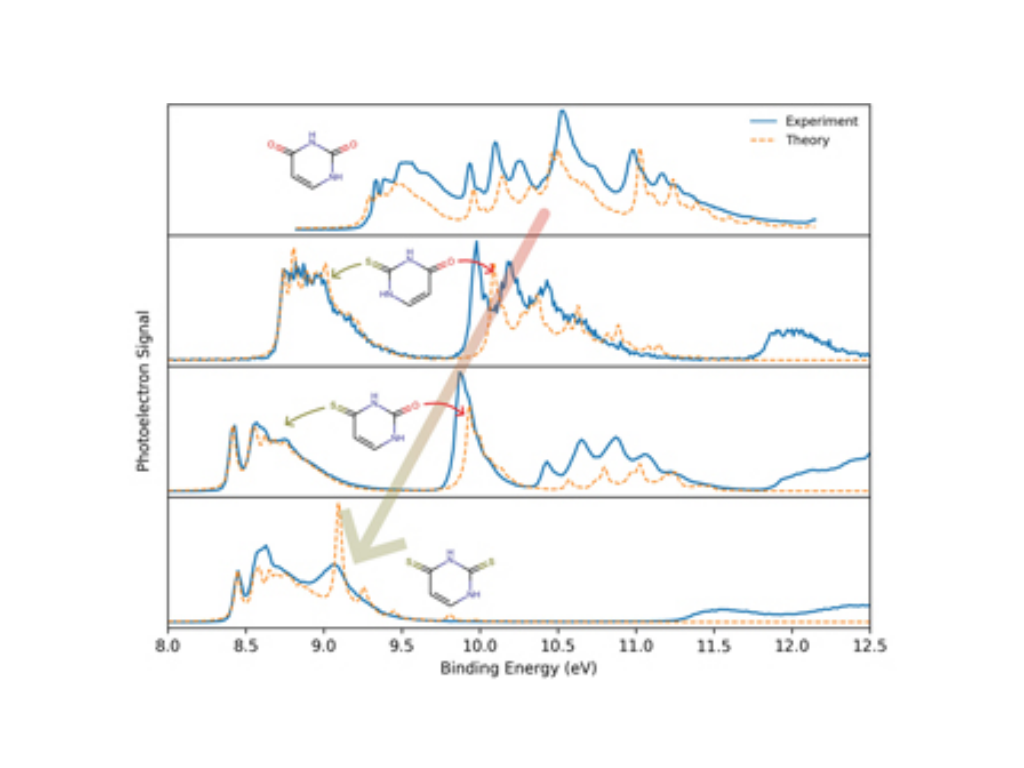
D. Mayer, E. Titov, F. Lever, L. Mehner, M. L. Murillo-Sánchez, C. Walz, J. Bozek, P. Saalfrank, M. Gühr
Valence photoelectron spectra of thiouracils in the gas phase
Journal of Chemical Physics 163, 074301 (2025)
We present a combined experimental and theoretical study of the vibrationally resolved valence photoelectron spectra of the complete series of thiouracils (2-thiouracil, 4-thiouracil, and 2,4-dithiouracil) for binding energies between 8 and 17 eV. The theoretical spectra were calculated using equation-of-motion coupled cluster theory for ionization potential combined with the time-independent double-harmonic adiabatic Hessian approach. For all three thiouracils, the first ionization potential is found between 8.4 and 8.7 eV, which is 1 eV lower than for the canonical nucleobase uracil. Ionization bands up to 12 eV show strong vibrational progressions and are well reproduced by the calculations. These bands are attributed to the ionization of (primarily) sulfur- and oxygen-localized valence molecular orbitals. For higher binding energies, the calculations indicate that nonadiabatic couplings are important for the interpretation of the photoelectron spectra.
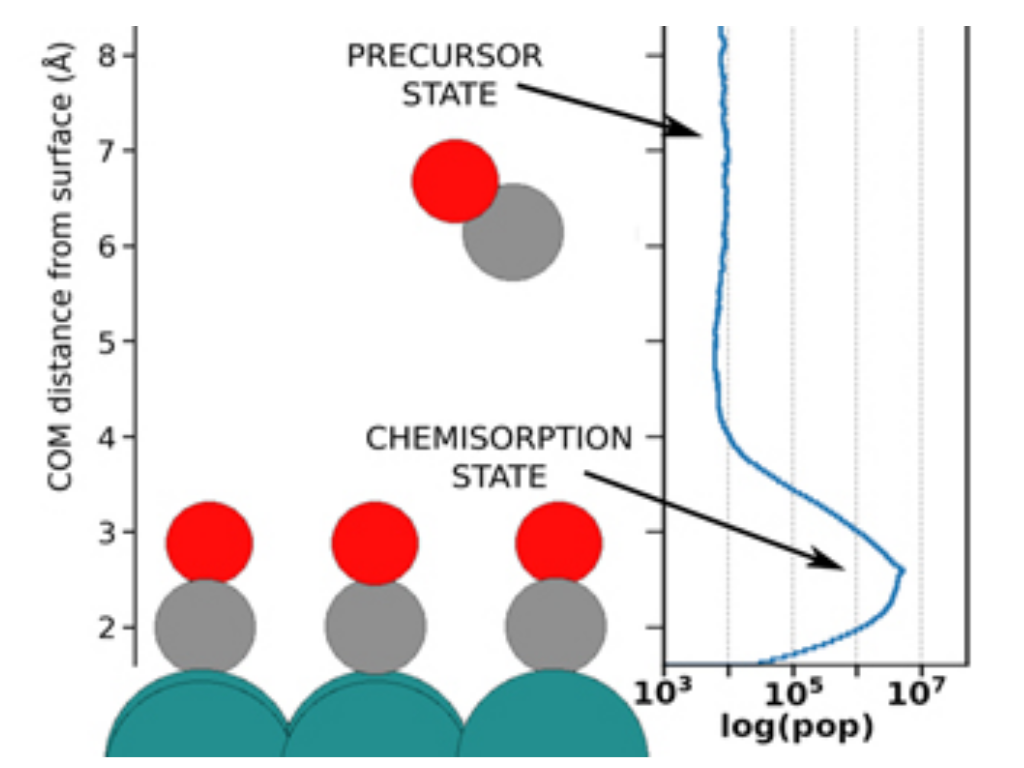
B. Mladineo, J. I. Juaristi, M. Alducin, P. Saalfrank, and I. Lončarić
Photoinduced dynamics of CO on Ru(0001): Understanding experiments by simulations with all degrees of freedom
Journal of Chemical Physics, 163, 044103 (2025)
Real-time pump–probe experiments are powerful tools for monitoring chemical reactions but often need parallel theoretical modeling to disentangle different contributions. Monitoring x-ray spectra of photoinduced dynamics of CO on Ru(0001) provided a strong indication for a transient “precursor state” of unidentified nature to various subsequent outcomes. So far, the precise nature of the postulated precursor has also remained elusive in state-of-the-art ab initio molecular dynamics models, including single-moving CO molecules. In the present work, we have constructed a density functional theory-based machine learning interatomic potential energy surface that is valid for all ionic degrees of freedom of the system, comprising many molecules at various coverages and moving surface atoms. Our Langevin dynamics with electronic friction based on the new potential energy surface identified the precursor state as dynamically trapped molecules around 6 Å from the surface that arise from adsorbate–adsorbate interactions. We have compared our results to experimental observations and calculated the dependence of reaction probabilities on pump laser fluence and initial surface coverage.
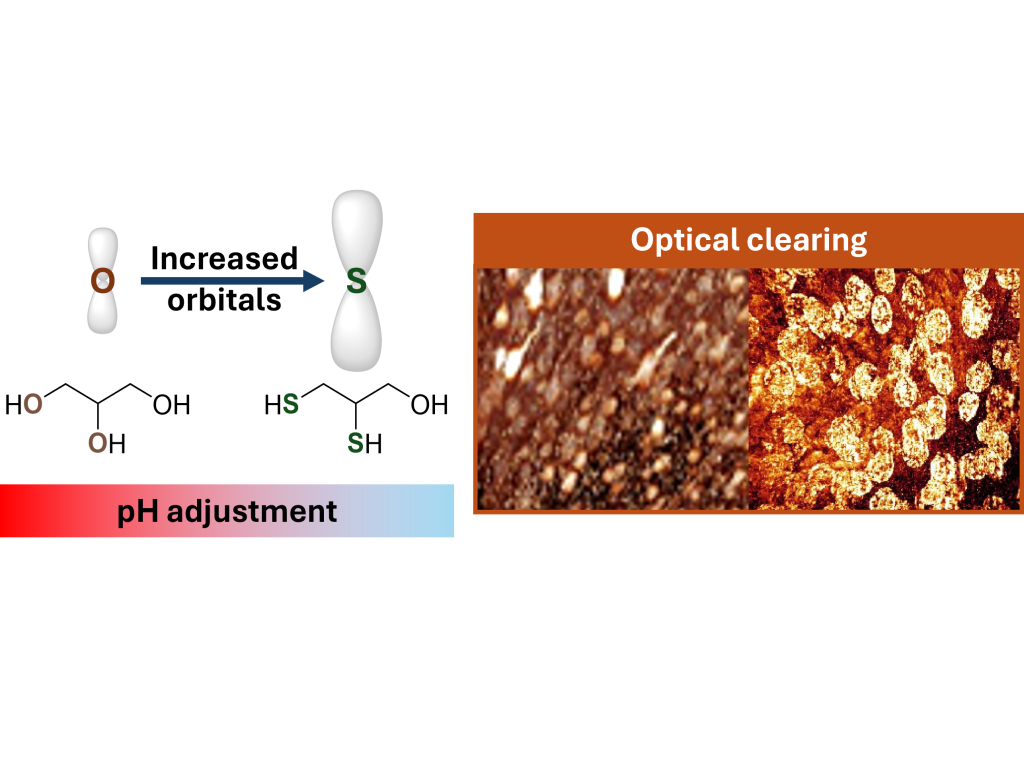
U. Leischner, M. Reifarth, M. S. Brill, F. Schmitt, S. Hoeppener, D. Unnersjö Jess, H. Brismar, U. S. Schubert, and R. Heintzmann
PRIAMOS: A technique for mixing embedding media for freely adjusting pH value and refractive index (RI) for optical clearing (OC) of whole tissue samples
Journal of Microscopy, 1–13, 2025
Investigations of biological samples often require sample transparency, which is achieved by embedding the sample in a high-refractive index (RI) medium to obtain a homogenous RI distribution in the sample, referred to as optical clearing (OC). Here, we introduce a method for designing embedding media with an increased RI by increasing molecular orbitals, which is achieved by replacing elements in molecules commonly used for OC with elements possessing a more pronounced polarisability. Briefly, we took the established embedding medium Glycerol and exchanged the OH-groups by Thiol-groups, resulting in an embedding medium with very similar properties, but with a higher refractive index. We describe a procedure—abbreviated PRIAMOS—to render biological samples transparent using an RI-matching liquid, which we refer to as pH-value and Refractive Index Adjustment by Mixing highly polarisable molecular Orbital Substances. We focus on optical clearing in three-dimensional tissue samples whilst preserving fluorescence of fluorescent labels. The clearing procedure requires 2–3 days, yielding highly transparent samples, preserving the fluorescence of fluorescent proteins like the yellow fluorescent protein (YFP). This is demonstrated on mouse brain samples, imaged with standard confocal microscopy down to 1.6 mm depth, as well as on kidney samples. Mixtures of monothioglycerol, dithioglycerol and tributylamine achieve RI values between 1.52 and 1.57, and an acidity equivalent to pH values between 5 and 8. Our PRIAMOS approach can serve as a guideline for optimising optical clearing protocols.
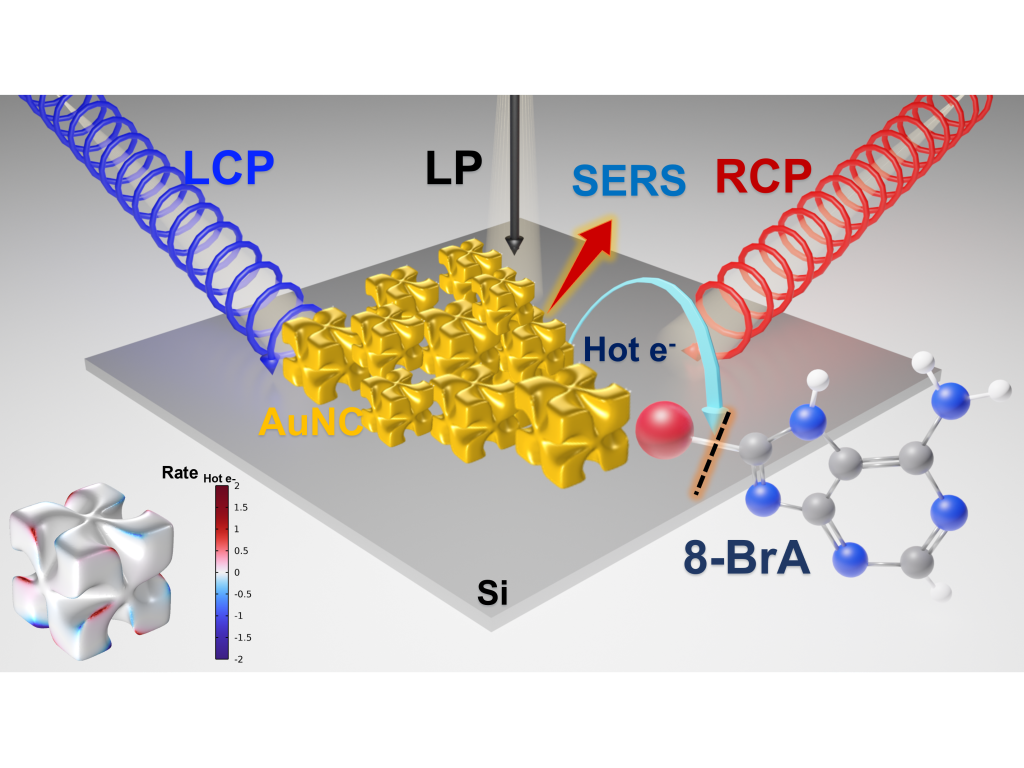
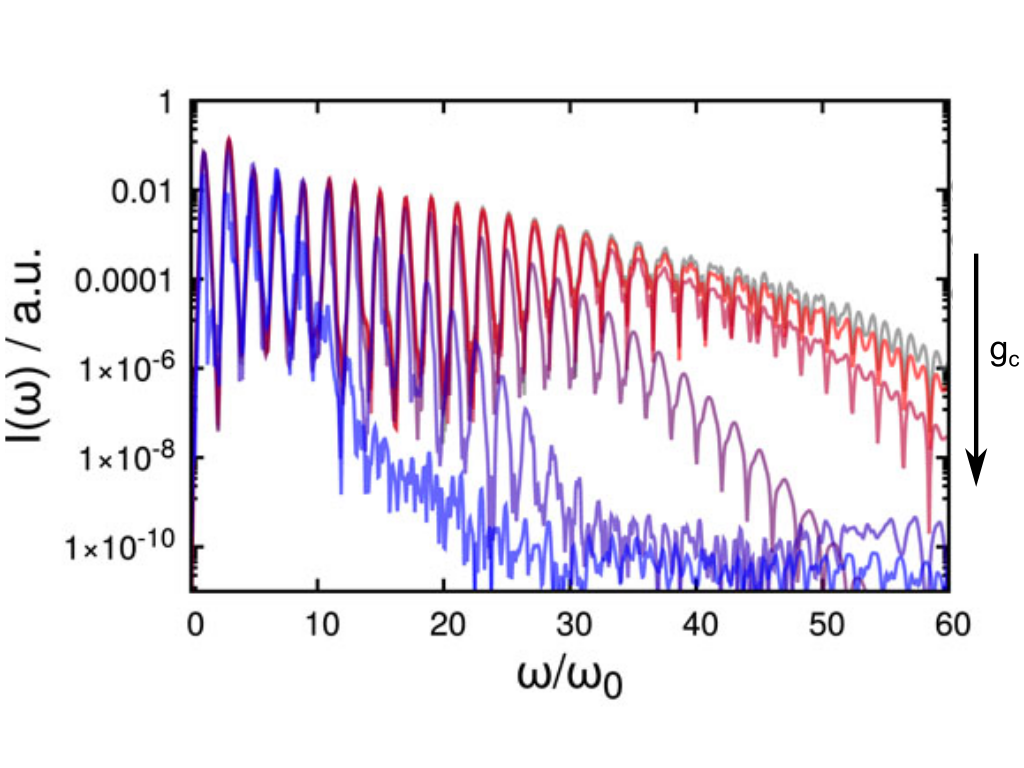
S. K. Gahlaut, O. Avalos-Ovando, R. Myeong Kim, R. Hussein, S. Juergensen, S. Reich, A. O. Govorov, K. Tae Nam, and I. Bald
Amplification of Photochemical Chiroptical Activity of Chiral Gold Nanocubes
Chiral plasmonic nanostructures enable exceptionally high dissymmetry factors (g-factors) compared to chiral molecules and present unparalleled opportunities in light manipulation, polarization-sensitive photochemistry, and chiral sensing. Here polarization-dependent plasmonic chemistry on chiral gold nanocubes (AuNCs) is presented, leveraging the high sensitivity of surface-enhanced Raman scattering (SERS). The AuNCs exhibit strong optical activity and localized surface plasmon resonances acting as highly efficient nanoscale light antennae. Employing the hot electron-induced dehalogenation of 8-Bromoadenine as a model reaction, it is demonstrated that circularly polarized light induces asymmetric reaction rates due to circular dichroism (CD) in hot electron generation efficiency. Astonishingly, the photochemical g-factor, quantified by the differential reaction rate coefficients under left-handed and right-handed circularly polarized light, surpasses its optical counterpart and can be further enhanced by laser intensity. Remarkably, multilayer assemblies of AuNCs exhibit a reversal in photochemical CD, which is tuneable via laser power and enables further g-factor enhancement. Comprehensive electromagnetic simulations of extinction spectra and hot electron generation maps corroborate the profound impact of particle arrangement on the optical g-factor and the g-factor for hot-electron generation. This work demonstrates a systematic approach to enhance the photochemical chiroptical response of chiral AuNCs, paving the way for extraordinary control over chemical reactions with light.
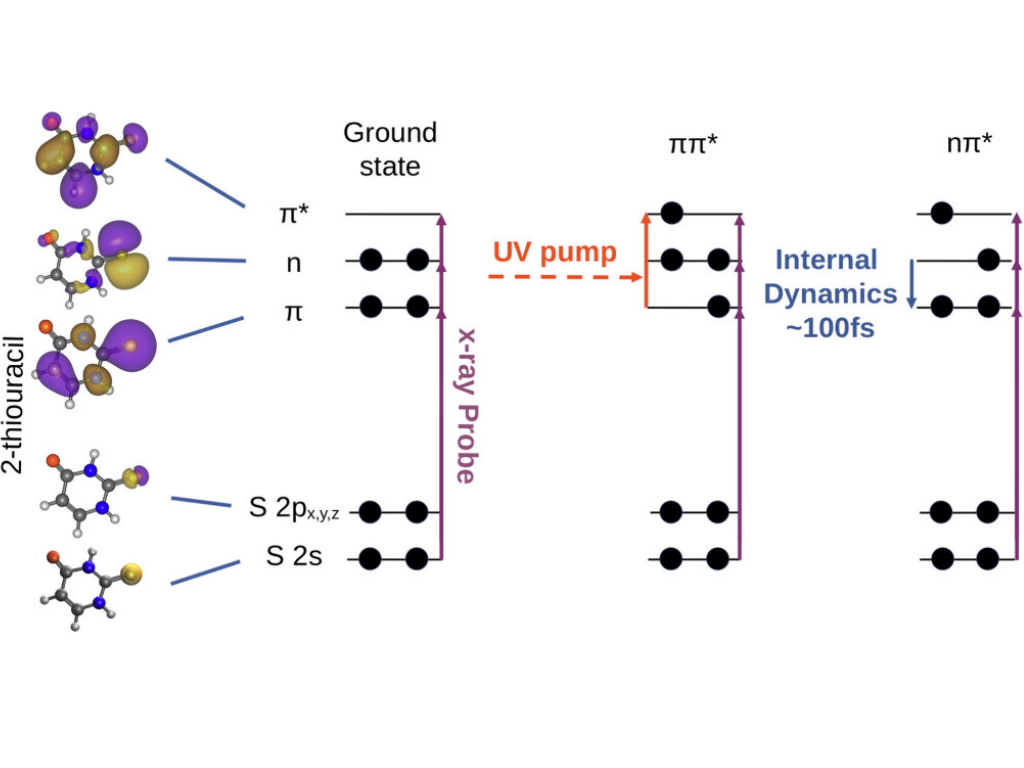
F. Lever, D. Picconi, D. Mayer, S. Ališauskas, F. Calegari, S. Düsterer, R. Feifel, M. Kuhlmann, T. Mazza, J. Metje, M. S. Robinson, R. J. Squibb, A. Trabattoni, M. Ware, P. Saalfrank, T. J. A. Wolf, M. Gühr
Direct Observation of the ππ* to nπ* Transition in 2-Thiouracil via Time-Resolved NEXAFS Spectroscopy
The Journal of Physical Chemistry Letters, 2025, 16, 16, 4038–4046
The photophysics of nucleobases has been the subject of both theoretical and experimental studies over the past decades due to the challenges posed by resolving the steps of their radiationless relaxation dynamics, which cannot be described in the framework of the Born–Oppenheimer approximation (BOA). In this context, the ultrafast dynamics of 2-thiouracil has been investigated with a time-resolved NEXAFS study at the Free Electron Laser FLASH. Near Edge X-ray Absorption Fine Structure spectroscopy (NEXAFS) can be used to observe electronic transitions in ultrafast molecular relaxation. We performed time-resolved UV-pump/X-ray probe absorption measurements at the sulfur 2s (L1) and 2p (L2/3) edges. We are able to identify absorption features corresponding to the S2 (ππ*) and S1 (nπ*) electronic states. We observe a delay of 102 ± 11 fs in the population of the nπ* state with respect to the initial optical excitation and interpret the delay as the time scale for the S2 → S1 internal conversion. We furthermore identify oscillations in the absorption signal that match a similar observation in our previous X-ray photoelectron spectroscopy study on the same molecule.

F Monticone, N. A. Mortensen, A. I. Fernández-Domínguez, Y. Luo, X. Zheng, C. Tserkezis, J. B. Khurgin, T. V. Shahbazyan, A. J. Chaves, N. M. R. Peres, G. Wegner, K. Busch, H. Hu, F. Della Sala, P. Zhang, C. Ciracì, J. Aizpurua, A. Babaze, A. G. Borisov, X.-W. Chen, T. Christensen, W. Yan, Y. Yang, U. Hohenester, L. Huber, M. Wubs, S. De Liberato, P. A. D. Gonçalves, F. J. García de Abajo, O. Hess, et al.
Nonlocality in photonic materials and metamaterials: roadmap
Optical Materials Express 15 (7), 1544-1709 (2025)
Photonic technologies continue to drive the quest for new optical materials with unprecedented responses. A major frontier in this field is the exploration of nonlocal (spatially dispersive) materials, going beyond the local, wavevector-independent assumption traditionally adopted in optical material modeling. The growing interest in plasmonic, polaritonic, and quantum materials has revealed naturally occurring nonlocalities, emphasizing the need for more accurate models to predict and design their optical responses. This has major implications also for topological, nonreciprocal, and time-varying systems based on these material platforms. Beyond natural materials, artificially structured materials—metamaterials and metasurfaces—can provide even stronger and engineered nonlocal effects, emerging from long-range interactions or multipolar effects. This is a rapidly expanding area in the field of photonic metamaterials, with open frontiers yet to be explored. In metasurfaces, in particular, nonlocality engineering has emerged as a powerful tool for designing strongly wavevector-dependent responses, enabling enhanced wavefront control, spatial compression, multifunctional devices, and wave-based computing. Furthermore, nonlocality and related concepts play a critical role in defining the ultimate limits of what is possible in optics, photonics, and wave physics. This Roadmap aims to survey the most exciting developments in nonlocal photonic materials and metamaterials, highlight new opportunities and open challenges, and chart new pathways that will drive this emerging field forward—toward new scientific discoveries and technological advancements.
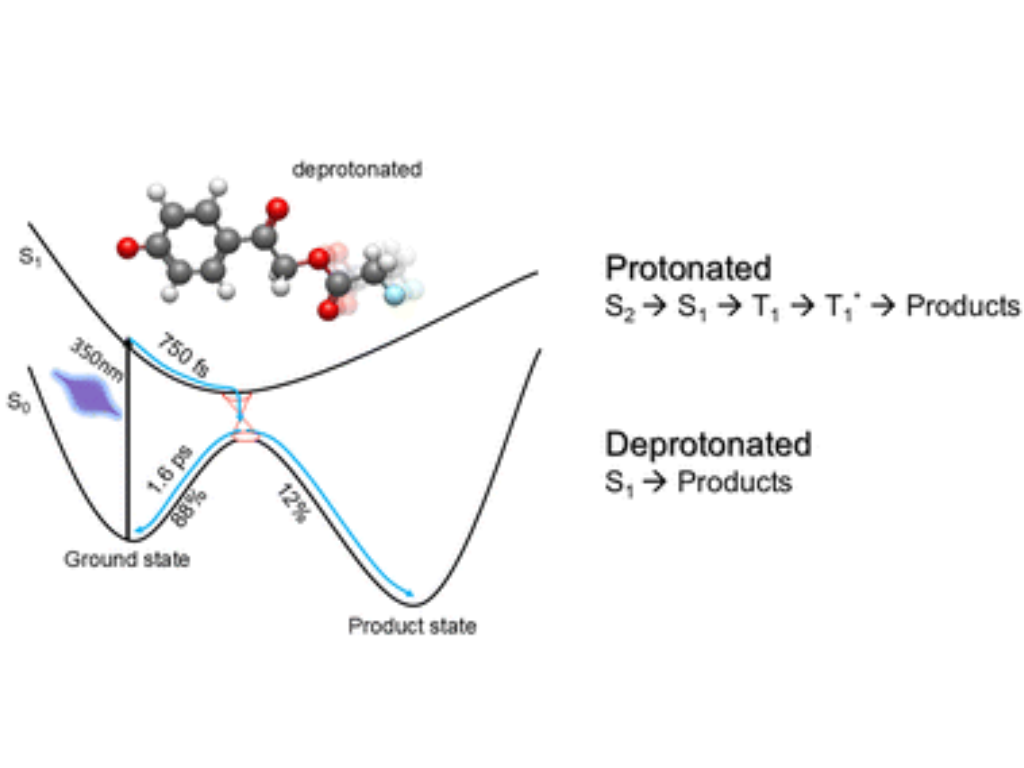
Y.Pfeifer, T. Stensitzki, J. Dostál, E. Titov, M. Kloz, P. Saalfrank and H. Müller-Werkmeister
pH-dependent ultrafast photodynamics of p-hydroxyphenacyl: deprotonation accelerates the photo-uncaging reaction
Physical Chemistry Chemical Physics, 2025, Advance Article
Photolabile protecting groups (PPGs) possess broad application potential as they allow spatially and temporally controlled release of the protected group. While the photochemistry of many PPGs has been studied in detail, data under aqueous conditions or depending on the pH are extremely rare. However, for applications under biological conditions in water, knowledge about the photochemistry under these is critical. Here, we studied the pH-dependent reaction dynamics of para-hydroxyphenacyl (pHP), one prominent example of PPGs, by ultrafast transient absorption spectroscopy and quantum chemical simulation. At neutral pH, the para-hydroxyl group of pHP is protonated and photoproduct formation occurs within less than 1 ns from a triplet state. At basic pH, the main scaffold gets deprotonated leading to a bathochromic shift of the characteristic absorption. Upon deprotonation the substrate release occurs directly from a singlet state, shortcutting the rate determining step of intersystem crossing (ISC) in the neutral case, resulting in an acceleration of the photochemical reaction with photoproduct formation observed at ∼1 ps.

V. Tkachenko, T. Wu, A. Heraji Esfahani, J. Gurke, M. Hartlieb
Choice of photo-excitation conditions in xanthate-supported photo-iniferter (XPI) RAFT polymerization
Polym. Chem., 2025, 16, 3070, 3078
The use of photo-controlled reversible deactivation radical polymerization (RDRP) techniques offers several advantages regarding the control of polymerization processes and material synthesis. Reversible addition-fragmentation chain-transfer (RAFT) polymerization is particularly interesting in this context as the chain transfer agent (CTA) used in these processes can be activated by light directly in a photo-iniferter (PI) process. We have recently introduced a method where a xanthate (a particularly active iniferter) is combined with a second CTA resulting in a xanthate supported (X)PI-RAFT process. Herein conducted DFT calculations suggest a significant difference in orbital contributions to the β-scission process when comparing xanthates and trithiocarbonates (TTC), offering an explanation for the improved performance of xanthate. Experimentally, we investigate how the choice of wavelength, light intensity and irradiation setup influences the control over the polymerization and in particular its livingness (end group fidelity). This is probed via the synthesis of multiblock and diblock copolymers where livingness is essential and can be accessed via deconvolution of the SEC traces. While a change in wavelength to more selectively activate the xanthate while not exciting the π-π* transition of the TTC does not seem to improve livingness, the light intensity was found to have a tremendous impact, with oversaturation being detrimental for chain end fidelity. Also using a flow-chemistry setup did not help to overcome this limitation. As such, the choice of light intensity was isolated as a tremendously important factor in XPI-RAFT polymerization.
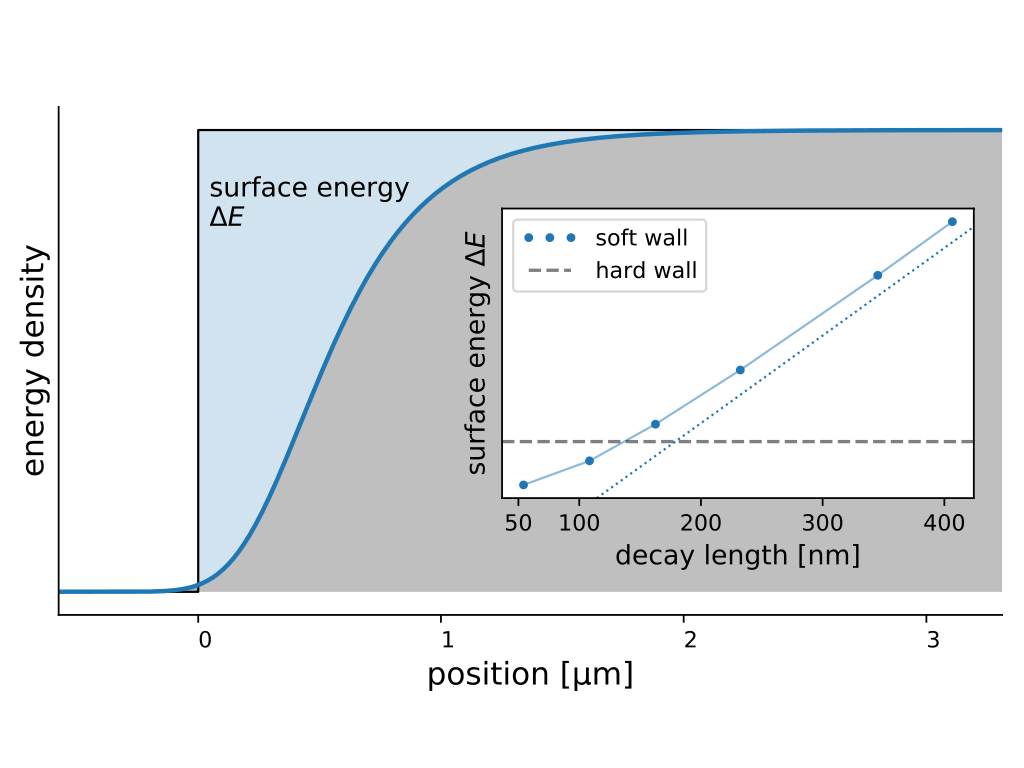
C. Henkel
Quantum borderlines – Fluctuation energies in ultracold Bose gases
Europhysics Letters, 150, 56001, 2025
Ultracold Bose gases are many-body systems with well-defined particle interactions that may serve as models for interacting quantum fields. The impact of virtual excitations is studied in the spatial transition zone created by a soft confinement potential that separates a degenerate ideal gas from a dense quasi-condensate. We compute within Bogoliubov theory the contribution of quasi-particles to the surface energy at zero temperature.
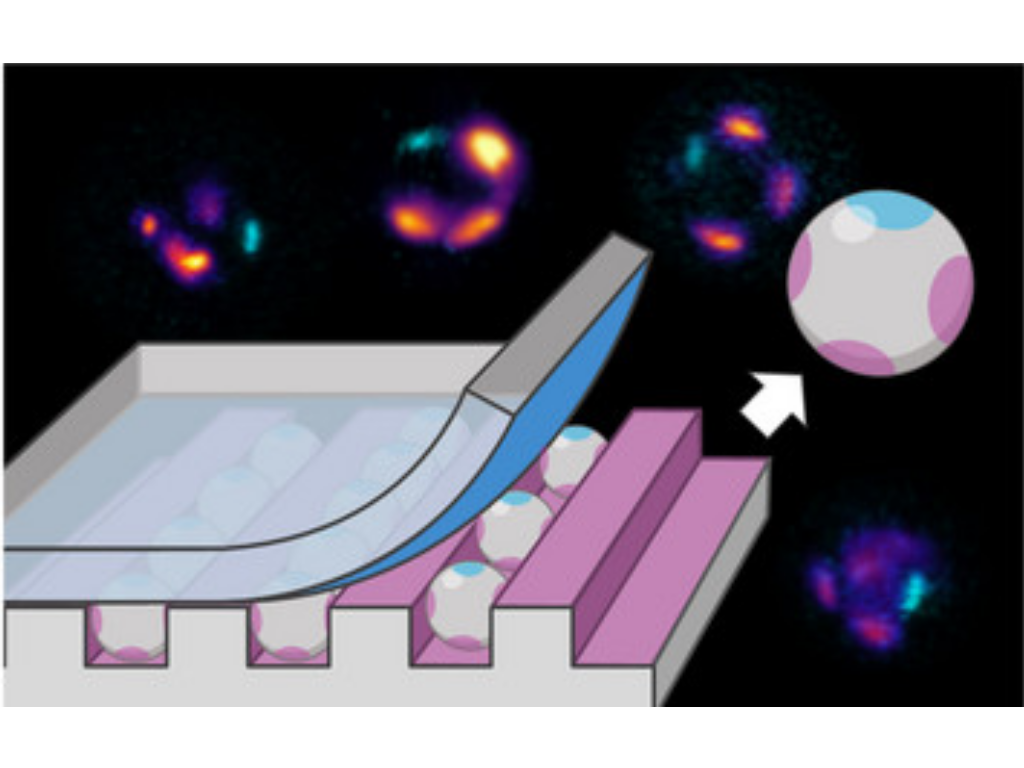
M. Reifarth, M. Sperling, R. Grobe, P. Akarsu, F. Schmitt, M. Schmette, S. Tank, K. M. Arndt, S. Chiantia, M. Hartlieb, A. Böker
Regioselective and Anisotropic Multi-Patching of Small Microparticles via a Polymer Brush-Assisted Microcontact Printing (µCP)
Advanced Functional Materials, 2025, 2423495
A method for fabricating anisotropic monodisperse silicon dioxide (SiO2) patchy microspheres is developed. In the protocol, we utilize a modified microcontact printing (µCP) technique, employing a polydimethylsiloxane (PDMS) elastomer with a regularly grooved surface topography as a stamp. The stamp's microscale channels create a confined environment for the SiO2 microspheres, as they match the particle dimensions (≈4 µm). The solid-phase µCP routine allows for the transfer of functional trialkoxysilanes from the stamp to the particles exclusively at their contact faces. By applying a second stamp to the exposed side of the particles, a fourth patch with different chemical information is added, resulting in particles with four equidistant patches along the particle equator. This fabrication process, compatible with a manifold follow-up chemistry, adds patches with adjustable characteristics. Consequently, a polymeric material containing the nucleobase adenine, is successfully added to the patches, which enables interactions with materials that possess its complementary base uracil. The method is suitable to fabricate particles possessing a C4v and C2v symmetry. Importantly, the protocol is easily adaptable to other particle dimensions and surface chemistry, possessing significant potential in the field of anisotropically functionalized spherical colloidal particles.
M. A. Beres, C. Boyer, M. Hartlieb, D. Konkolewicz, G. G. Qiao, B. S. Sumerlin and S. Perrier
RAFT with Light: A User Guide to using Thiocarbonylthio Compounds in Photopolymerizations
ACS Polymers Au 2025, 5, 3, 184–213
This perspective offers an in-depth guide to photopolymerizations mediated with thiocarbonylthio compounds, with a particular focus on photoiniferter and photoinduced energy/electron transfer RAFT (PET-RAFT) polymerizations, focusing on practical considerations. It is designed to provide both newcomers and experts with the practical knowledge needed to harness light-mediated polymerizations for innovative applications. The discussion begins with an overview of conventional RAFT polymerization and proceeds to highlight the distinctive advantages of the photomediated processes. The photochemical behavior of thiocarbonylthio compounds, along with the selection of appropriate light wavelengths, is critically examined for its impact on polymerization kinetics and optimization of polymer properties. Key parameters influencing polymerization success─such as catalyst selection, solvent choice, light intensity, and temperature─are explored in detail. The importance of oxygen tolerance and end-group fidelity is also addressed, as these factors are essential for achieving well-defined polymers. Additionally, reactor configurations are reviewed, focusing on the roles of light sources, reactor geometry (batch versus flow systems), and temperature control in optimizing the reaction efficiency. The article concludes by integrating these concepts into a comprehensive framework for optimizing photoiniferter and PET-RAFT polymerizations.
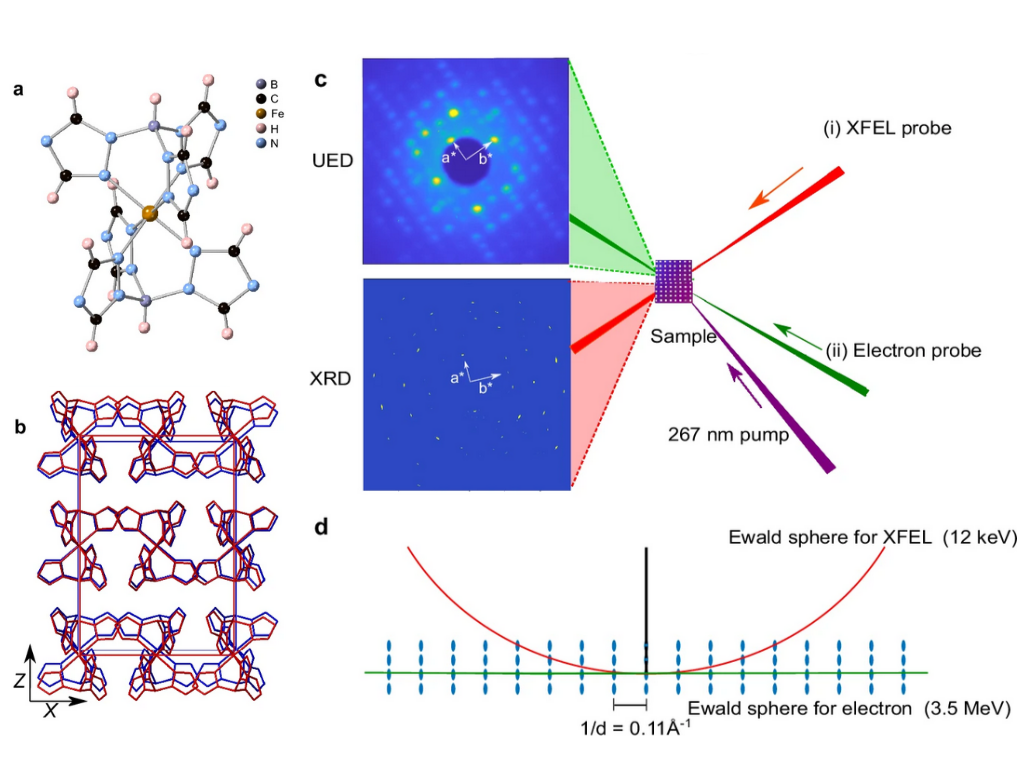
D. Vinci, K. Ridier, F. Qi, F. Ardana-Lamas, P. Zalden, L. Chung Liu, T. Eklund, M. Sielemann Jakobsen, R. Schubert, D. Khakhulin, C. Deiter, N. Bottin, H. Yousef, D. von Stetten, P. Łaski, R. Kamiński, K. N. Jarzembska, R. F. Wallick, T. Stensitzki, R. M. van der Veen, H. Müller-Werkmeister, G. Molnár, D. Xiang, C. Milne, M. Lorenc and Y. Jiang
Capturing ultrafast molecular motions and lattice dynamics in spin crossover film using femtosecond diffraction methods
Nature Communications, 2025, 16, 2043
A comprehensive insight into ultrafast dynamics of photo-switchable materials is desired for efficient control of material properties through light excitation. Here, we study a polycrystalline spin crossover thin film as a prototypical example and reveal the sequential photo-switching dynamics, from local molecular rearrangement to global lattice deformation. On the earliest femtosecond timescale, the local molecular structural rearrangement occurs within a constant unit-cell volume through a two-step process, involving initial Fe−ligand bond elongation followed by ligand rotation. The highly-oriented structure of the nanocrystalline films and the experimental geometry enables resolving the full anisotropic lattice structural dynamics in and out of the sample plane separately. While both molecular switching and lattice heating influence lattice volume, they exert varying degrees of impact at disparate time scales following photoexcitation. This study highlights the opportunities provided by Mega-electron-volt electron and X-ray free electron laser to advance the understanding of ultrafast dynamics of photo-switchable materials.
Book Chapters

E. W. Fischer, P. Saalfrank
A theoretical chemistry approach to vibro-polaritonic chemistry with application to infrared spectroscopy and reaction kinetics
The emerging interdisciplinary research field of vibro-polaritonic chemistry exploits the concept of vibrational strong coupling (VSC) to shape chemical reactivity and molecular properties. Vibro-polaritonic chemistry employs optical Fabry–Pérot cavities as a novel light source, which provide access to VSC between confined infrared (IR) radiation modes and molecular (ro)vibrational degrees of freedom. VSC induces the formation of light–matter hybrid states known as vibrational polaritons, which are experimentally characterized by a paradigmatic doublet signature in linear IR spectra. Mechanistically even more intriguing is the experimentally reported observation of VSC-modified ground state chemistry. From a conceptual perspective, vibro-polaritonic chemistry differs from traditional laser-based light–matter interaction scenarios: While the latter commonly rely on a semiclassical approach subject to a classical description of the electromagnetic field, in vibro-polaritonic chemistry the entire light–matter hybrid system is described quantum mechanically. This chapter provides a contemporary overview of vibro-polaritonic chemistry from the perspective of a theoretical chemist. Theoretical concepts extending the common quantum chemical perspective towards molecular interactions with quantized cavity radiation fields are presented in an introductory fashion. Applications to linear IR spectroscopy and reaction kinetics in the VSC regime are illustratively discussed for selected model problems.