Articles
R. M. Sarhan, H. H. Srinivasalu, S. von Chamier Gliszczinski, S. Kogikoski Jr., Yan Lu, B. Schmidt, and I. Bald
Nonthermal Charge Carriers Control Selectivity in Plasmon-Induced Suzuki–Miyaura Cross-Coupling Reaction on Spiked Au/Pd Nanowires
ACS Catalysis 2026, XXXX, XXX, XXX-XXX
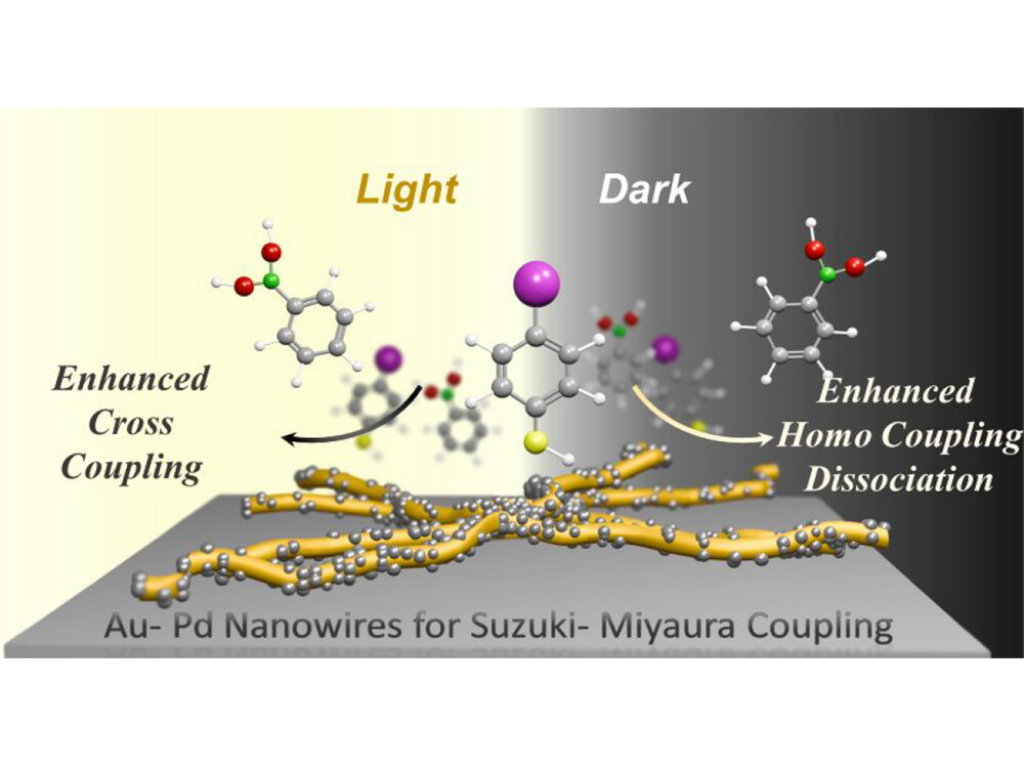
Palladium (Pd)-catalyzed reactions, such as the Suzuki–Miyaura cross-coupling, have demonstrated significant enhancement when facilitated by hybrid Au/Pd nanostructures. However, the role of gold, through charge transfer or localized heating, remains debated. Moreover, the impact of plasmon excitation on reaction selectivity remains largely unexplored. Herein, we report a facile, two-step, room-temperature synthesis of spiked Au/Pd nanowires for visible-light-driven Suzuki–Miyaura cross-coupling under ambient conditions. The reaction was monitored in situ using liquid-state surface-enhanced Raman spectroscopy (SERS) and ex situ using gas chromatography-mass spectrometry (GC-MS). We systematically studied the cross-coupling of 4-iodothiophenol with phenyl boronic acid under varying excitation wavelengths, laser intensities, external heating, and charge carrier scavengers, revealing the distinct role of non-thermal charge carriers in selectively promoting formation of the biaryl product. In the dark, a temperature of 85 °C was required to initiate the reaction, while thiophenol was observed as a major by-product. To further validate the plasmon-induced selectivity, we performed the reaction with 4-iodotoluene entirely in solution and monitored the products using GC-MS. Under light irradiation, cross-coupling was favored over the thermally driven reactions in the dark. This highlights the role of non-thermal charge carriers in enhancing the selectivity of Suzuki cross-coupling under plasmonic excitation.
J.-E. Pudell, M. Mattern, F. Baltrusch, F. L. Boariu, M. Kronseder, R. Shayduk, A. Madsen, M. Rössle, M. Bargheer, D. Schick, and A. von Reppert
Ultrafast x-ray thermometry: Contrasting strain and Debye-Waller effects in platinum thin films
Physical Review B, 113, L220301, 2026
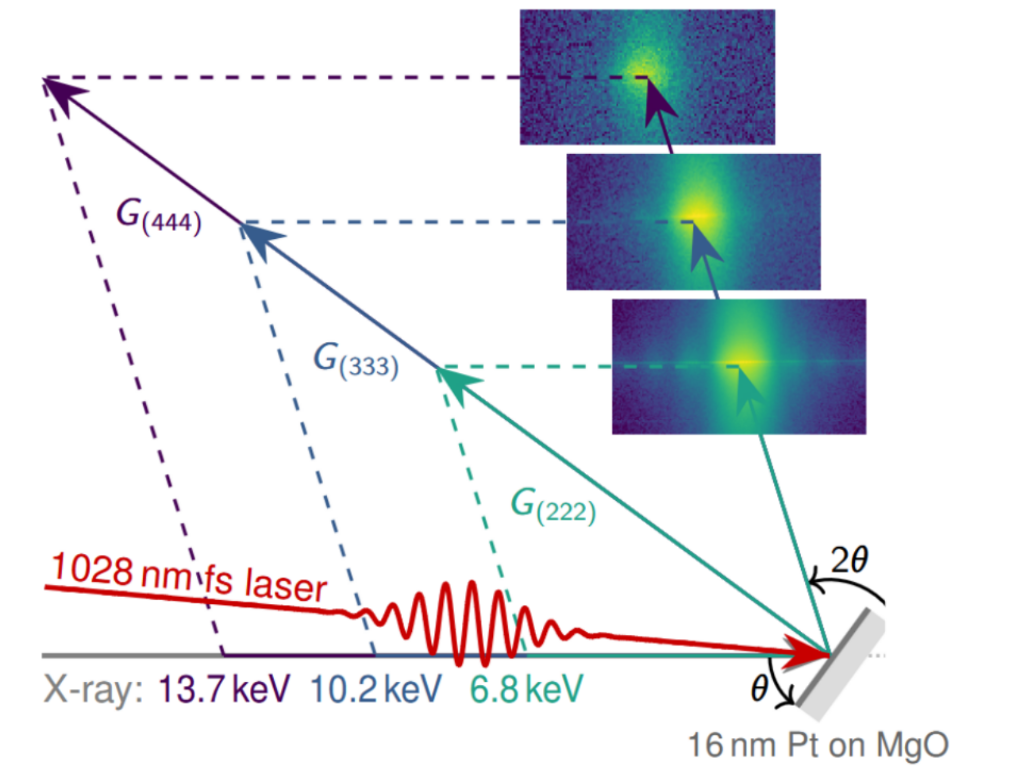
Thermal energy and temperature govern a wide range of physical properties and dynamics in solids. X-ray diffraction can be used to monitor material-specific temperatures on ultrafast timescales and within nanostructures by exploiting thermal expansion or the Debye-Waller effect. Here, we simultaneously track the intensity and position changes of different out-of-plane Bragg peaks of a 16nm thick Pt film on an MgO substrate upon equilibrium heating and upon femtosecond laser excitation to quantify the mean-square atomic displacement and the lattice expansion. Our comparison of these two x-ray thermometers experimentally verifies that the in-plane expansion of homogeneously excited continuous single-crystalline thin films is forbidden on picosecond timescales. This drastically changes the out-of-plane thermal expansion coefficient, i.e., the relationship between the observed lattice expansion and the corresponding temperature increase. Thus, considering the boundary conditions of in-plane lattice expansion is generally indispensable for extracting temperatures from the lattice expansion in diffraction experiments.
N. Jahn, and E. Titov
Understanding Molecular Excited States at the Metal–Molecule Interface via Transition Density Matrix Analysis─A Case Study of Azobenzene Thiols on Gold
ACS Physical Chemistry Au 2026, XXXX, XXX, XXX-XXX
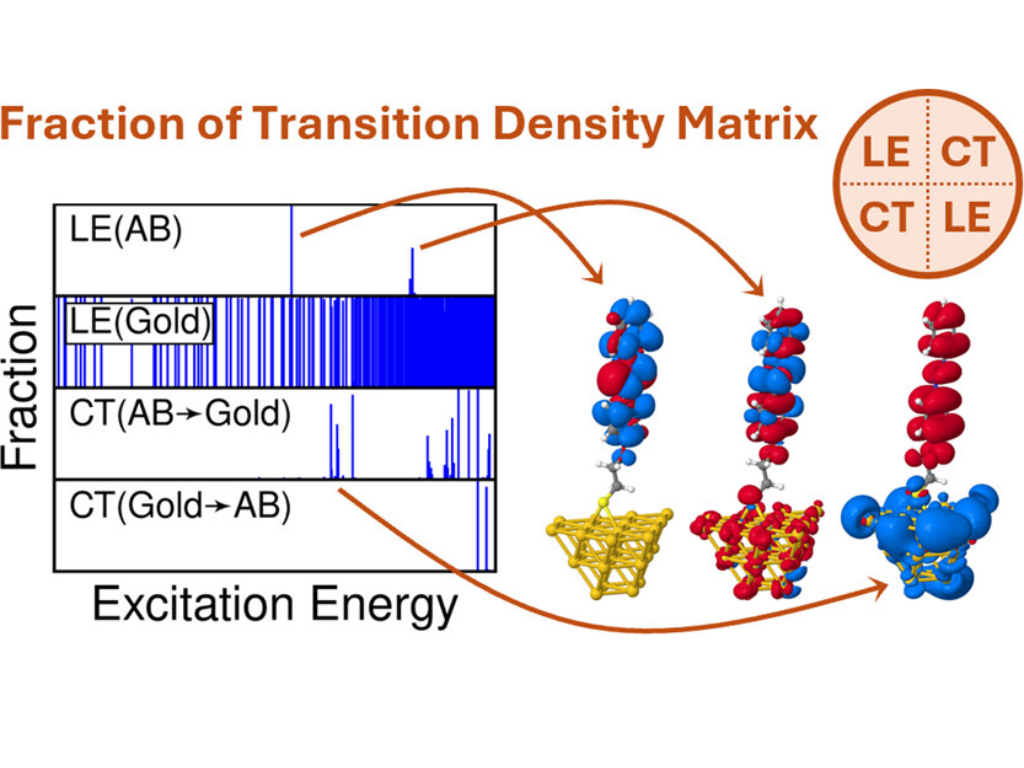
Noble metal nanoparticles are rapidly gaining popularity as novel catalytic platforms to influence chemical reactions in various ways. Despite an increasingly large number of studies, many key processes especially at the metal–molecule interface are fundamentally still not fully understood up to this day. Throughout this work, we present a systematic study targeting the molecular excited states of covalently linked azobenzenes (ABs) on gold via a combination of time-dependent density functional theory and tight-binding calculations with transition density matrix analysis. We find that the optically bright ππ* state is strongly resonant to close-lying local gold excitations, which leads to splitting and redshift of the ππ* state and increased UV/vis absorption around the ππ* excitation energy. We show that these findings hold true across different AB isomers and demonstrate how a systematic variation of the AB–gold distance leads to a gradual localization of the ππ* excitation. Moreover, we discuss how a direct electronic interaction between the AB and the gold surface leads to the formation of delocalized hybrid states and investigate the exciton formation of AB dimers at the metal interface. Our results are carefully verified across a wide range of computational parameters including a large number of different density functionals, basis sets, and gold clusters of varying forms and sizes. The presented workflow is easily applicable to other functional molecules on metal surfaces to further broaden the understanding of substrate–surface interactions at the interface.
A. Zehle, C. Penschke, and E. Titov
Molecular excitons in arylazopyrazole aggregates: a quantum chemical study
Scientific Reports, 16, 18029 (2026)
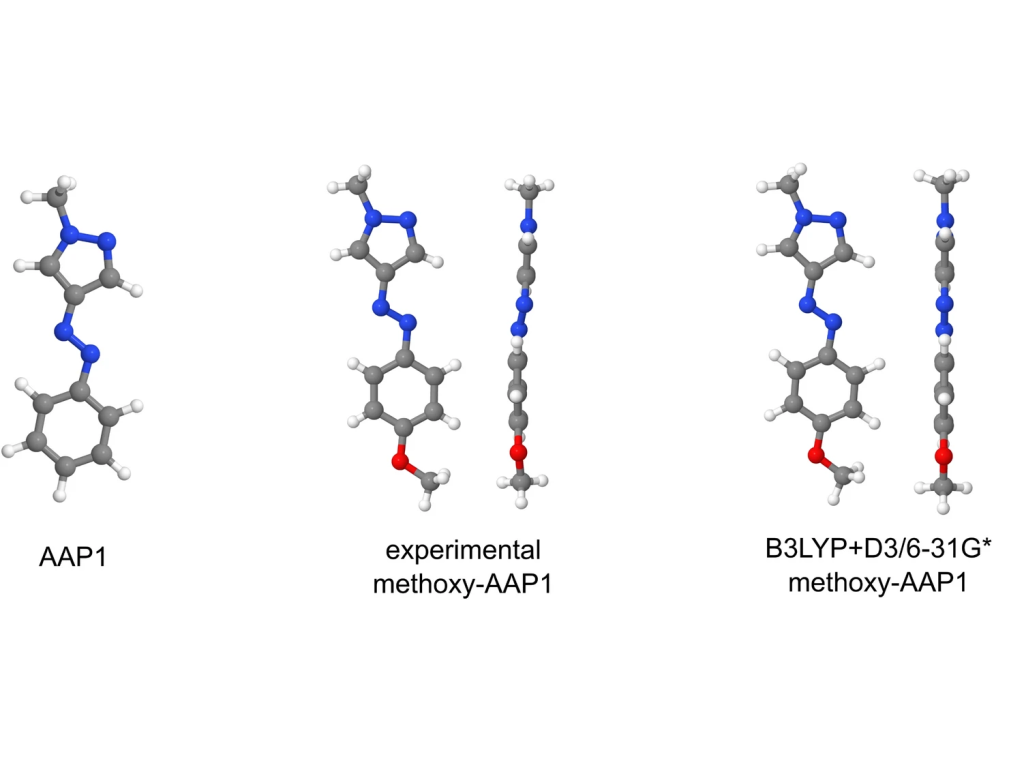
Aggregation of molecular photoswitches may affect their functionality. Fundamentally, interaction of monomers in the aggregated state results in formation of exciton states, which, in turn, govern energy and charge transfer processes in the materials made of the photoswitches. In this work, we study the exciton states of aggregates of arylazopyrazole — the photoswitch which gained popularity in last decade as an alternative to azobenzene — using quantum chemical calculations. We perform cluster excited-state calculations for aggregates including up to 32 arylazopyrazole monomers as well as periodic calculations for the crystal structure. We obtain and analyze the composition of the exciton states, exciton splittings, and monomer-to-aggregate spectral shifts, thus providing quantitative insight into the electronic states and absorption spectra of realistic arylazopyrazole aggregates.
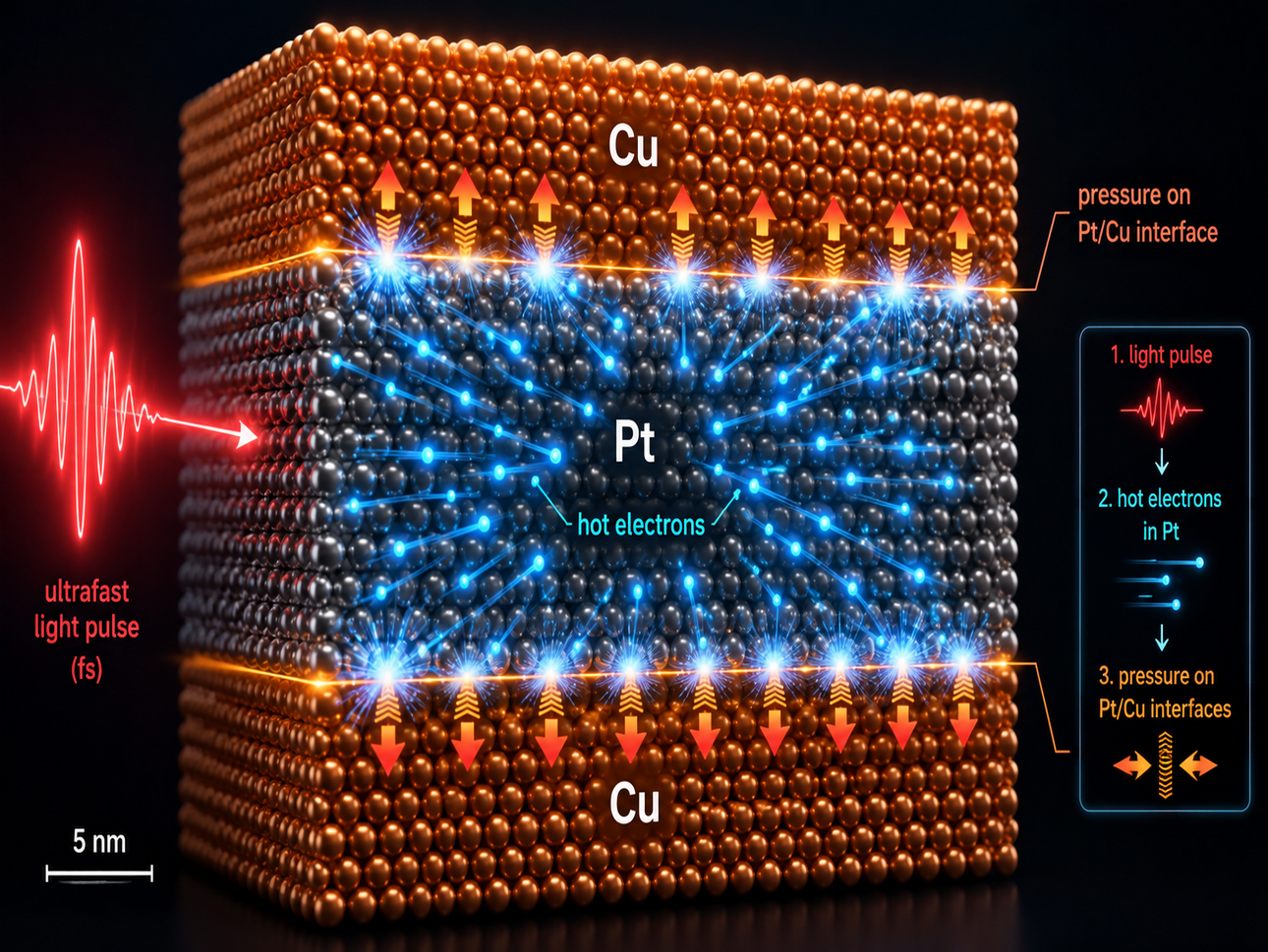
J.-E. Pudell, M. Mattern, M. Herzog, A. von Reppert , C. K. Singh, D. Schick, M. Hehn, U. Boesenberg, A. Rodriguez-Fernandez, R. Shayduk, W. Jo, J. Möller, J. Hallmann, J. Wrigley, P. M. Oppeneer, A. Madsen, and M. Bargheer
Electron pressure drives THz phonons in metal-metal superlattices
Nature Communications 17, 5308 (2026)
Ultrafast control of lattice motion in metals is a central challenge for highfrequency strain engineering and spintronic applications. Coherent strain control at terahertz (THz) frequencies in metals has remained elusive because free electrons are expected to delocalize energy beyond the optical penetration depth, preventing rapid and efficient stress generation.Here we show that robust and cost-effective metal–metal superlattices (SLs), where periodic repetitions of bilayers — each layer a few atoms thick — are deposited by
sputtering, constitute thermoacoustic metamaterials that overcome this limitation. We combine femtosecond X-ray diffraction with mode-resolved density-functional theory and two-temperature modeling to show that electron pressure, rather than phonon stress, drives a large-amplitude coherent terahertz (1 THz) lattice oscillation in sputtered Pt/Cu superlattices. We establish electron pressure as an engineerable, dominant actuation mechanism in metallic metamaterials which can be tailored by the pitch and the constituent
materials of the sputtered SL structure, enabling applications such as ultrafast strain-mediated antiferromagnetic spintronic devices.
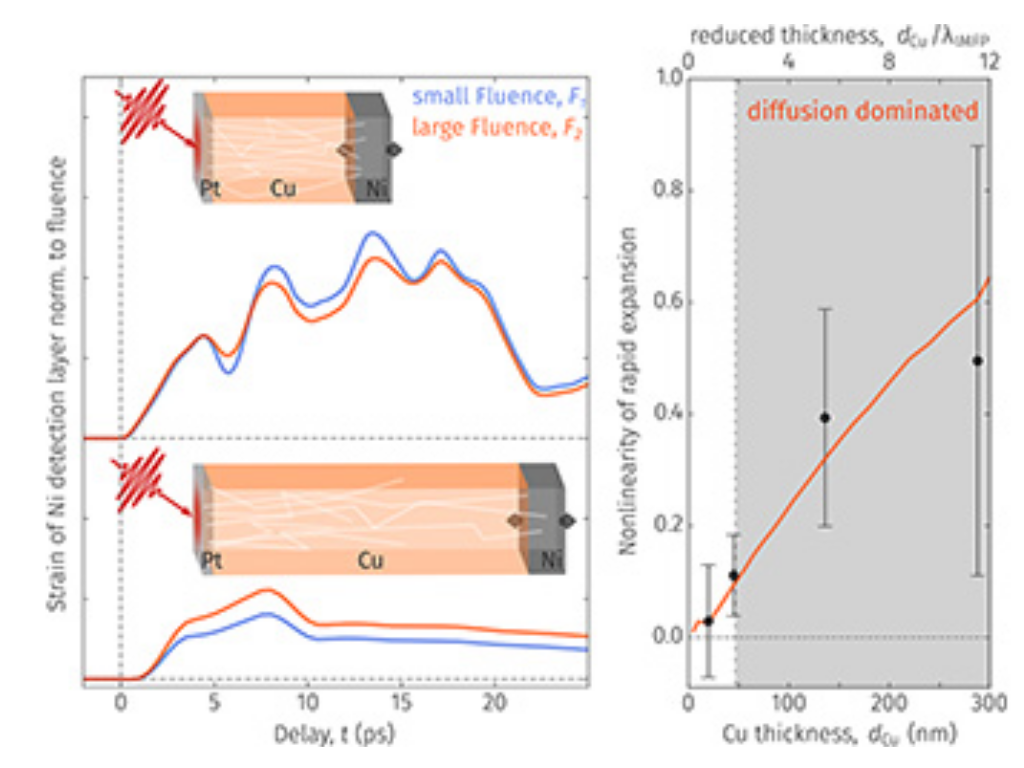
J. Jarecki, L. Mehner, M. Mattern, A. Jurgilaitis, S. P. Zeuschner, B. Ahn, F. Baltrusch, J. C. Ekström, D. Kroon, M. Herzog, C. Walz, F.-C. Weber, J. Larsson, M. Hehn, J.-E. Pudell, D. Schick, A. von Reppert, M. Bargheer
Experimental evidence of dominsnt ultrafast diffusive energy transport by hot electrons in Cu
Applied Physics Letters 128, 172204 (2026)
When the dimensions of structures shrink to the order of the inelastic mean free path of the energy-carrying quasi-particles, the character of energy transport changes from diffusive to ballistic. However, the point of transition remains a matter of debate. Here, we leverage the fluence-dependent transport efficiency to distinguish ballistic and diffusive electron transport in an approach not relying on the transport velocity. We follow the energy that is rapidly transferred across Cu layers of different thicknesses via hot electrons from a photo-excited Pt layer into a buried Ni detection layer. In the Ni layer, the transported energy linearly relates to a rapid lattice expansion, which we probe via ultrafast x-ray diffraction. A nonlinear dependence of the Ni strain amplitude on the absorbed laser fluence indicates that the transport through Cu becomes more efficient with increasing fluence, which is inconsistent with a ballistic scenario but reproduced by a diffusive energy transport model. We already identify that for a Cu thickness of about 50 nm, i.e., about twice the electronic inelastic mean free path, diffusive electronic energy transport dominates the spatial energy distribution. Our experimental approach is generally applicable to distinguish diffusion from ballistic energy transport.
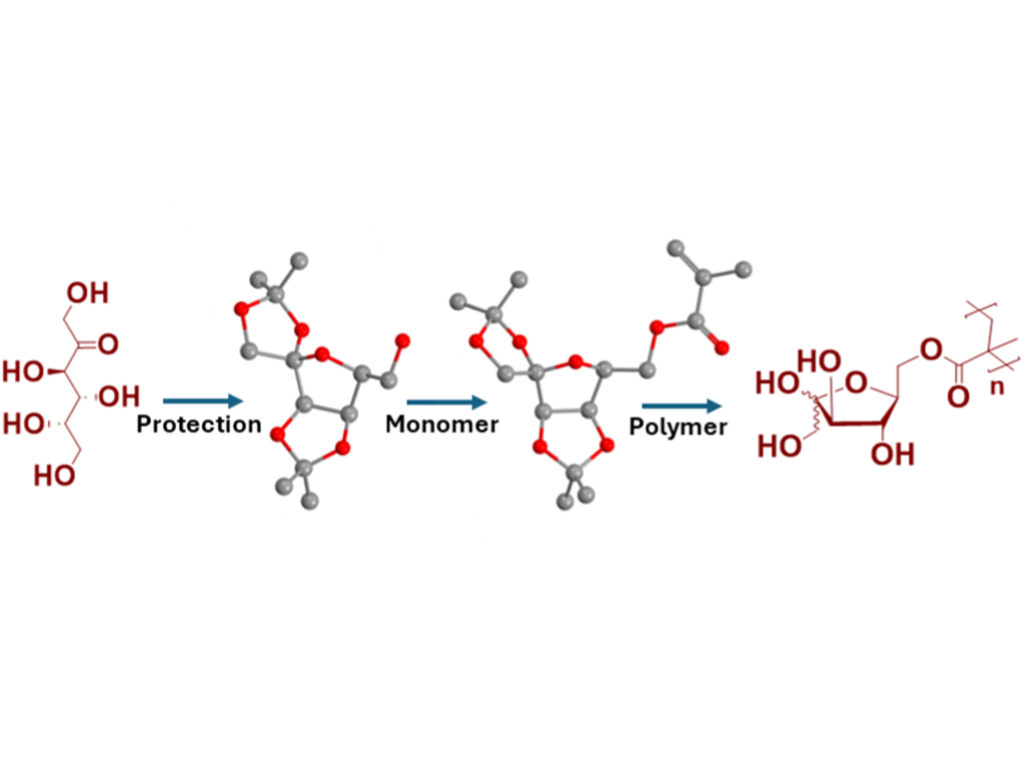
N. Pallab, M. Schenderlein, E. Titov, E. Sperlich, M. Schmette, and M. Reifarth
D-Fructose, L-Sorbose, D-Tagatose, D-Psicose: Functional Methacrylate-Based Glycopolymers of Ketohexoses Possessing Enhanced Boronic Acid Affinity
The unique binding properties of ketohexoses with boronic acids present new opportunities for functional material design. We investigated the binding of D-fructose, L-sorbose, D-tagatose, and D-psicose with four boronic acids (phenylboronic acid (PBA), 3-acetamidophenylboronic acid (AcPBA), benzoxaborole (BOB), and 6-acetamidobenzoxaborole (AcBOB)) in aqueous environment using isothermal titration calorimetry (ITC), finding that D-psicose exhibits exceptional affinity, compared to other ketohexoses. The ketohexose sugars were efficiently converted into isopropylidene-protected methacrylate monomers for polymer formation. These polymers can be deprotected in an acidic condition to yield the side-chain functionalized glycopolymers, providing a versatile platform for strong boronic acid–carbohydrate interactions for dynamic and advanced material design.

A. Nabiyan, M. Esfandiari, S. Kogikoski Jr., I. Bald, E. Titov, M. U. Kumke, N. Kulak, and H. Schlaad
Polyelectrolyte Copolymer Nanoreactors: From Colloidal Assembly to Photoredox Activity in Water
ACS Applied Materials & Interfaces 2026, 18, 13 19622-19634
Performing organic photoredox reactions in water remains challenging because most catalysts cannot simultaneously solubilize substrates, control nanoscale organization, and maintain activity under aqueous conditions. We report a photoredox-active polyelectrolyte based on a polydehydroalanine (PDha) backbone covalently functionalized with polypyridyl complexes to address some of these limitations. The copolymer undergoes substrate-triggered self-assembly in water, forming photocatalytically active spherical colloidal nanostructures (∼30 nm), as confirmed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The assemblies efficiently catalyze the hydroxylation of arylboronic acids, a representative water-insoluble photoredox transformation. Mechanistic studies using UV-visible spectroscopy, Raman spectroscopy, time-resolved emission spectroscopy, electrochemistry, and density functional theory (DFT) indicate that dual hydrogen bonding between PDha carboxylates and arylboronic acids governs both self-assembly and catalytic performance. The nanostructures retain high activity over multiple cycles. These findings establish adaptive polymer self-assembly as a general strategy for creating enzyme-like, water-compatible photoredox systems and provide a platform for transferring organic photoredox chemistry into aqueous media.
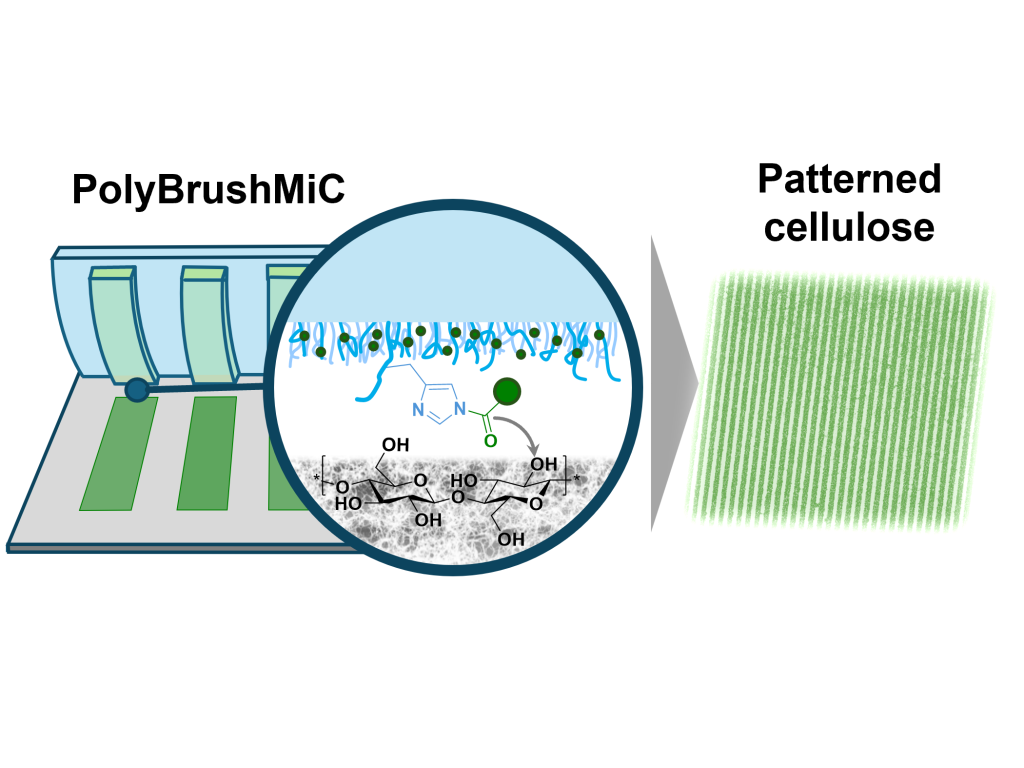
N. Pallab, M. Schmette, S. Kogikoski, Jr., K. Hettrich, M. Schenderlein, and M. Reifarth
(Sub-) Microscale Structuring of Cellulose Thin Films using a Polymer Brush-Assisted Microcontact Printing (PolyBrushMiC) Routine
We present a method for directly micropatterning cellulose thin films using microcontact printing with a polymer-brush stamp carrying reactive imidazole groups. These temporarily bind carboxylic-acid inks, enabling clean ester-forming transfer to cellulose without smearing. This allows high-resolution, chemical patterning, paving the way for the fabrication of functional papers in a simple and efficient process.

B. Bhattacharyya, C. Balischewski, J. Kim, T. Duarte, L. Fu, R. A.S. Ferreira, A. Beqiraj, I. Mikulska, D. Gianolio, E. Sperlich, F. Stete, W. Koopman, C. Günter, K. Brennenstuhl, D. Van Opdenbosch, C. Zollfrank, A. Wedel, B. J. Murray, M. Swadźba-Kwaśny, T. Klamroth, M. U. Kumke, V. de Zea Bermudez, and A. Taubert
Halide-Dependent Photoluminescence and Heavy-Atom Effects in Low-Melting Organic–Inorganic Manganese Halides
Advanced Functional Materials (2026): e32016
Low-melting ionic solids with stirring luminescent properties hold significant promise for optoelectronic applications. Here, we compare and contrast the structural and spectroscopic correlations of two highly luminescent organic-inorganic manganese halides (C4Py)2[MnCl4] and (C4Py)2[MnBr4], synthesized from their respective manganese halides and N-butyl pyridinium halide ionic liquids. Although both compounds exhibit very similar bulk structures (determined by single-crystal and powder X-ray diffraction) and overall similar electronic structures (as indicated by the density of states), they differ notably in their optical properties. The chloride salt, (C4Py)2[MnCl4], has a photoluminescence decay lifetime ten times longer than its bromide analogue, (C4Py)2[MnBr4]. Furthermore, PL-quantum yield of (C4Py)2[MnBr4] is 1.6 times higher than that of (C4Py)2[MnCl4], which was attributed to the heavy atom effect of bromine atoms, based on periodic density functional calculations (with and without spin-orbit coupling). Although photoluminescence is only exhibited in the solid state, EXAFS analysis confirms that the coordination environment of manganese is remarkably similar in crystalline and molten states, potentially suggesting that photoluminescence is associated with the long-range crystalline order, which is lost upon melting. Building on these fundamental studies, the potential of (C4Py)2[MnCl4] as a luminescent security ink for anticounterfeiting applications has been demonstrated.
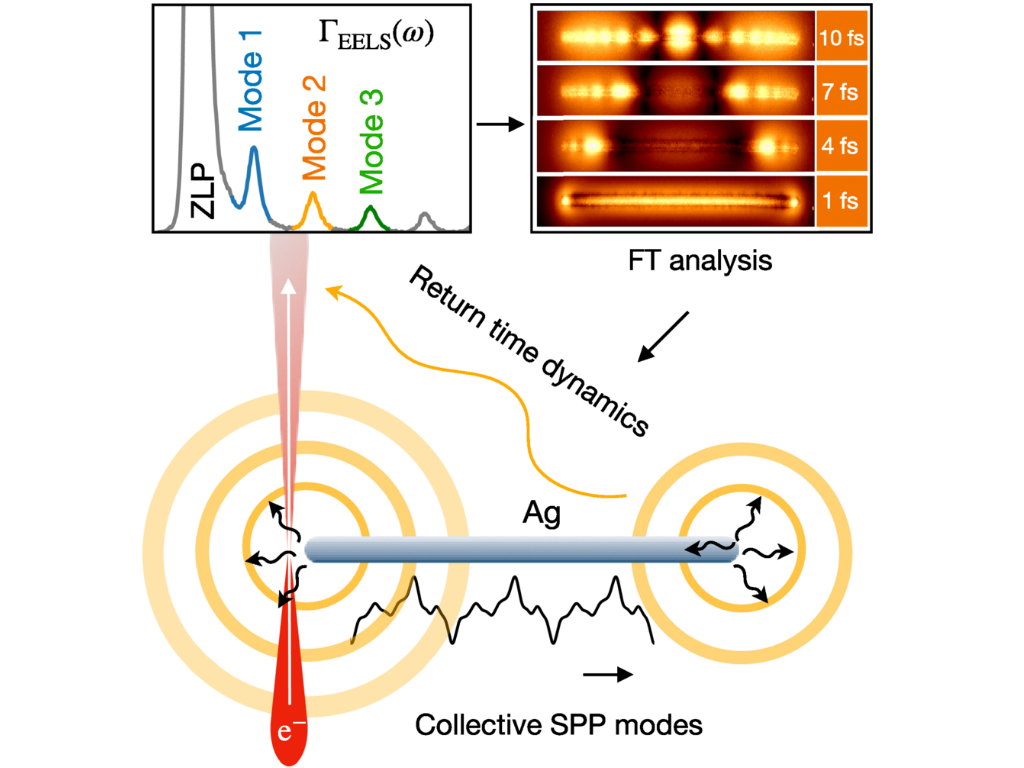
W. Zhao, Á. Rodríguez Echarri, A. Eljarrat, H. C. Nerl, T. Kiel, B. Haas, H. Halim, Y. Lu, C. T. Koch, and K. Busch
Time-domain study of surface plasmon polariton propagation in silver nanowires
Physical Review B 113, 085425 (2026)
Electron microscopy techniques such as electron energy-loss spectroscopy (EELS) facilitate the spatiospectral characterization of plasmonic nanostructures. In this work, a time-dependent perspective is presented that significantly enhances the utility of EELS. In particular, this approach facilitates the analysis of the dynamics of plasmonic excitations that repeatedly interact with swift electrons in a STEM-EELS configuration. This includes the bulk plasmon mode, which can only be excited by penetrating electron beams, and the fundamental surface plasmon polariton modes propagating along the wire, which can be excited by both penetrating and aloof trajectories. In addition, the role of higher-order azimuthal surface plasmon polariton modes, often overlooked for very thin wires, is observed and analyzed in both the energy-loss spectrum and from the dynamical perspective. Such a complete understanding of the interaction of electrons and plasmonic excitations is key for the design of efficient plasmonic sensors, the study of hot electron dynamics in metals, and applications in the context of electron quantum optics, where full control of the spatial and temporal characteristics of the fields at the nanometer and femtosecond scales is highly desirable.

L. Cordsmeier, W. Ribeiro da Silva Neto, M. Fondell, R. Mitzner, V. Vaz da Cruz, S. Eckert, and A. Föhlisch
Mechanisms of Hydroxyl Radical Chemistry in Aqueous Solution Triggered by Photoexcitation and Probed by Soft X-rays
Journal of the American Chemical Society , 2026, 148, 8, 8567–8573
Hydroxyl radicals are among the most important radicals on earth, being present in the human body, the atmosphere, rivers, and oceans, contributing to mechanisms like oxidative stress in cells and the photochemistry of the troposphere, and posing a threat to aquatic life. Extensive use of fertilizers in agriculture has led to increased levels of nitrogen oxides in many rivers around the world, which are a major source of hydroxyl radicals in water. In this paper, we explore the photoinduced generation of hydroxyl radicals from nitrite and their scavenging by the radical scavenger 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) in aqueous solutions using transient soft X-ray absorption spectroscopy (XAS) at the oxygen and nitrogen K-edges. We show the photoinduced generation of hydroxyl radicals from nitrite and determine its mechanism. For the scavenging of hydroxyl radicals by TEMPO, we show that the mechanism does not proceed through a bound intermediate state between the two molecules, as has been proposed in the literature, but instead through an electron transfer.
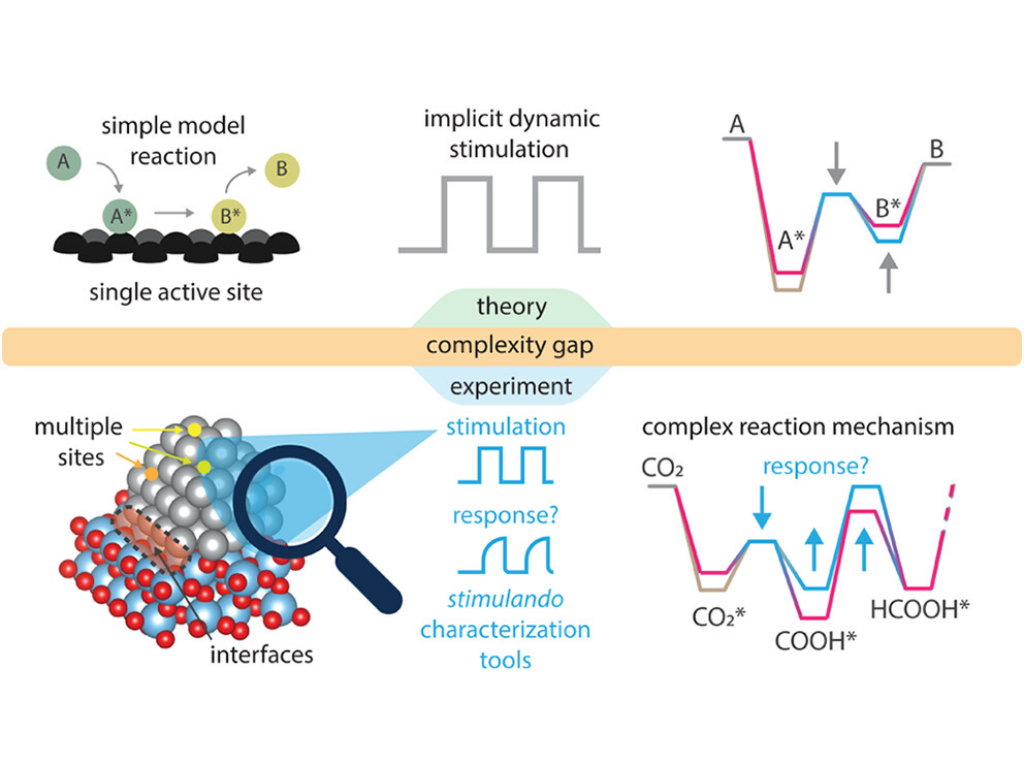
M. Monai, W. Albrecht, A. Alkemper, N. Artrith, A. Baldi, A. Beck, R. T. Berry, E. Bianco, F. A. Brzesowsky, Q. Dong, J. Faria Albanese, R. R. Frontiera, E. Galvin, E. C. Garnett, N. Gerrits, M. Grzelczak, M. Herzog, F. Hess, A. A. Kolganov, W. Koopman, N. Kosinov, S. Lander, E. Lepre, D. N. Maaskant, G. Miao, A. M. Naik, T. M. Onn, A. A. Peterson, D. Piankova, E. A. Pidko, K. Trangwachirachai, F. van den Bosch, D. Xu, B. Yilmaz, J. Zeininger, E. Alarcón Lladó, J. Meyer, P. J. Dauenhauer, S. H. C. Askes
Grand Challenges and Opportunities in Stimulated Dynamic and Resonant Catalysis
ACS Catalysis 16, 4077 (2026).
Traditional heterogeneous catalysis is constrained by kinetic and thermodynamic limits, such as the Sabatier principle and reaction equilibrium. Dynamic and resonant catalysts hold promise to overcome these limitations by actively oscillating a catalyst’s physical or electronic structure at the time scale of the catalytic cycle, allowing programmable control over reaction pathways, and leading to improved rate and selectivity. External stimuli such as temperature swing, mechanical strain, electric charge, and light can perturb catalyst surfaces in different ways, altering adsorbate coverage, binding energies, and transition states beyond what steady-state catalysis allows. This work surveys the current state of dynamic catalysis, introduces the concept of “stimulando” characterization for observing transient dynamics, and outlines key modeling, mechanistic, and benchmarking strategies to advance the field toward improved chemical transformation.
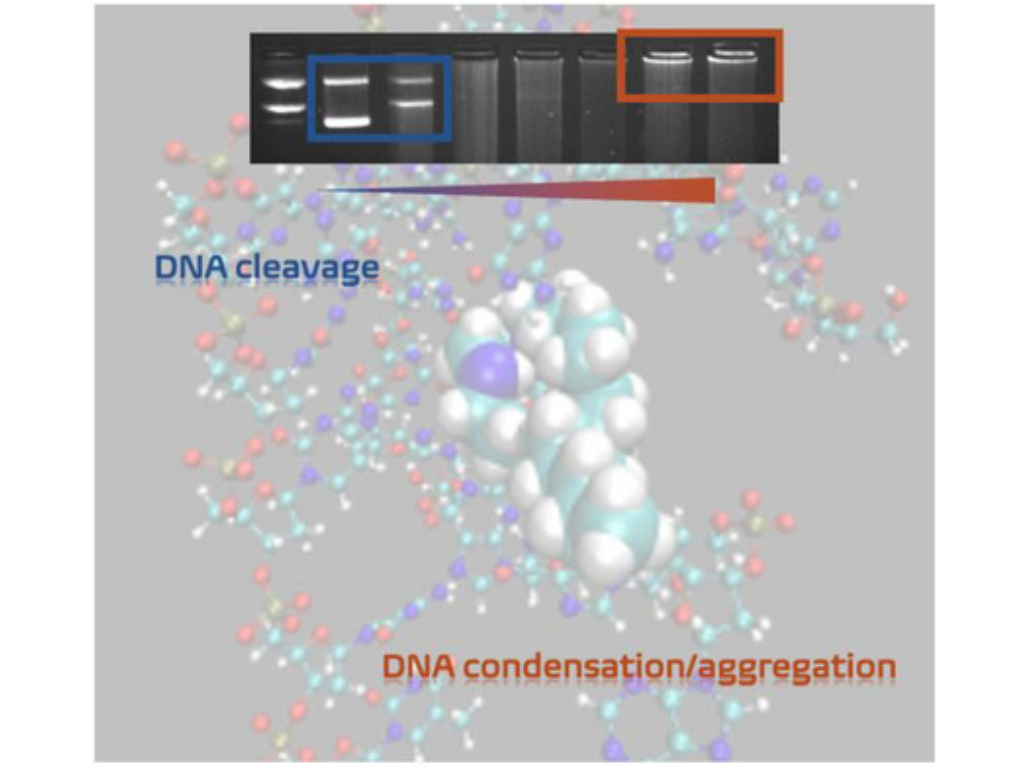
O. Verbitsky, S. Hinojosa, A. Mostafa, D. Ojha, I. Bald, and N. Kulak
Amphiphilic Cu(II) Oxacyclen Complexes: From Oxidative Cleavage to Condensation of DNA
ChemBioChem 2026, 27, e202500477
Cu(II) complexes with monoalkylated oxacyclen ligands (C12, C16, and C18) have been investigated regarding their interaction with DNA by different methods: circular dichroism, UV/VIS (ultraviolet-visible) and fluorescence spectroscopy as well as by gel electrophoresis. The results demonstrate that the complexes can cleave DNA through both hydrolytic and oxidative mechanisms, with hydroxyl radicals and hydrogen peroxide identified as the reactive oxygen species involved. The targeted incorporation of alkyl chains significantly enhances the DNA-binding affinity of the Cu(II) complexes, and the length of the alkyl substituents plays an important role, as they can interact with the major groove of the DNA. Alkylation is the determining structural factor responsible for the enhanced DNA interaction, since such an interaction is not observed with unsubstituted complexes. Moreover, the length of the alkyl chains significantly influences this behavior, as longer substituents induce a concentration-dependent DNA aggregation, a phenomenon absent in the nonalkylated analog. This aggregation and condensation behavior is examined using atomic force microscopy and dynamic light scattering. Moreover, DNA/small molecule interactions are also investigated using molecular dynamics simulations.
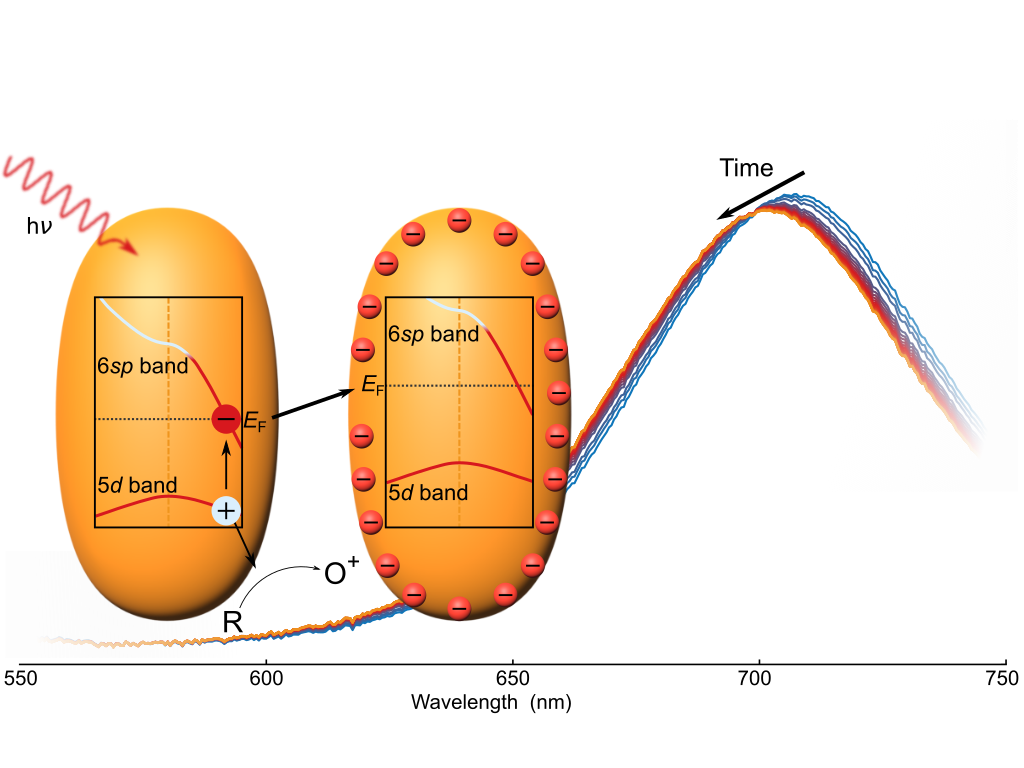
F. Stete, M. Bargheer, and W. Koopman
Capacitive photocharging of gold nanorods
Nature Communincations 17, 139 (2026)
Light can charge plasmonic nanoparticles by photoredox reactions, significantly modifying their optical and chemical properties. However, the charging process has been challenging to track experimentally, severely hindering its thorough evaluation. In this study, we investigate the charging of gold nanorods during a light-induced reaction in situ, utilizing the sensitivity of the rods’ longitudinal localized surface plasmon resonance to charge accumulation. Describing the particles as nanocapacitors, we present a model to quantify the number of charges on the particles and their connection to the illumination intensity. We find that the Fermi level, together with all other energy bands, is raised because of the repulsive potential of the additional charges. Experimental observations of the dependence on the solvent, the particle size, and ligand type further corroborate the proposed capacitor model. The results presented in this study lay the groundwork for the rational engineering of dynamic charge accumulation during plasmon-driven photoreactions.
Preprints
S. Kogikoski Jr, W. Koopman, F. Stete, R. M. Sarhan, S. K.Gahlaut, E. Titov, M. Bargheer, and I. Bald
Molecule–Metal Nanostructure Interactions in Plasmonic Chemistry
Plasmonic chemistry converts metal nanostructures into photocatalysts via three interconnected channels at the molecule–metal interface: enhanced near fields, non-equilibrium charge carriers, and localized heat. This review focuses on this interfacial perspective. We show how adsorbates and ligands do more than just stick to nanoparticles – they reduce plasmon lifetimes, change work functions, modify the density of states, and impact the energy and charge flow during various physical processes. Ultrafast thermalization under pulsed excitation and steady state conditions under continuous illumination open up different pathways for chemical reactions. Although the plasmonic metal is crucial, surface-bound molecules actively participate by scavenging charge carriers, affecting selectivity and stability, and even undergoing chemical transformations. These aspects are crucial for controlling product distributions and enabling novel chemical pathways by exploiting excited-state pathways inaccessible through thermal energy. Together with the nanoscale properties of plasmonic nanostructures, chemical reactions can be confined to nanoscale hot spots shaped by geometry and polarization. These insights inform the development of practical design principles for antenna–reactor hybrids and novel plasmonic materials in which absorption, carrier localization, and interfacial alignment are precisely engineered. Finally, we identify key open questions and the need for operando interfacial techniques to transform these insights into practical tools for selective, solar-driven synthesis.
B. Roy, E. Titov, and P. Saalfrank
A Theoretical Investigation of Nonadiabatic Dynamics and Time-Resolved NEXAFS Spectra of Uracil
Uracil, a fundamental RNA nucleobase, exhibits complex excited-state dynamics governed by nonadiabatic processes that prevent photodamage. Upon photoexcitation, population initially reaches the bright ππ∗ (S2) state and subsequently relaxes through the dark nπ∗ (S1) state via conical intersections, before returning to the ground state on sub-to few-picosecond timescales. In this work, we investigate these relaxation pathways using time-dependent density functional theory (TD-DFT)-based nonadiabatic surface hopping dynamics in combination with simulated time-resolved near-edge X-ray absorption fine structure (TR-NEXAFS) spectroscopy of oxygen, nitrogen and carbon K-edges. This combined approach allows us to capture both electronic and nuclear motion during the ultrafast relaxation and to identify atom-specific contributions to the decay process. In particular, ππ∗ to nπ∗ transitions occurring within the first 150 fs leave traces in O 1s and C 1s NEXAFS signals, indicating C=O bond elongations and torsional motion along the C=C bond, respectively. Overall, our results provide a detailed picture of population transfer, electronic-state trapping, and structural evolution in photoexcited uracil.